
PFAS: wait-and-see Vs precautionary principle
When data is limited, do you default to safety or wait for proof of harm? This post explores the widening regulatory gap between the US and EU regarding PFAS - the "Forever Chemicals."
If you had to decide whether something is safe based on limited data, which way would you default?
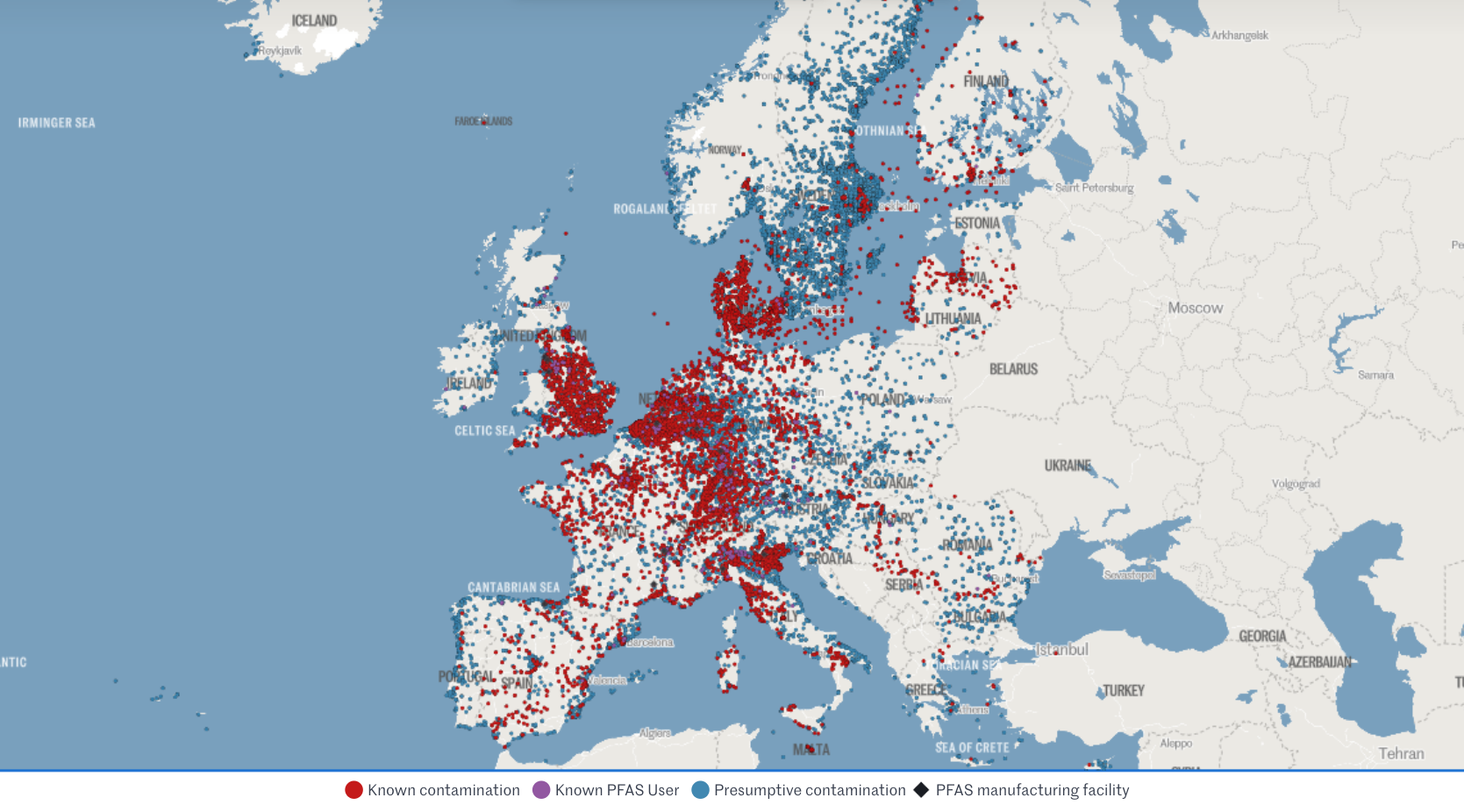
Let's look at recent regulatory developments re "Teflon-like" chemicals (PFAS) in cosmetics and medical devices. Per- and polyfluoroalkyl substances (PFAS) are highly inert synthetic chemicals which makes them sought after for both everyday uses and specialist ones. However, they are so inert that biology cannot break them down. They persist in the environment and accumulate in creatures at the top of the food chain: us.
The regulatory approach to PFAS, also called Forever Chemicals, is another staggering example of the US vs. EU cultural divide.
U.S. wait-and-see approach
🇺🇸 Context: In 2024, FDA launched the Modernization of Cosmetics Regulation Act (MoCRA) which required registration of all cosmetics and listing of all their ingredients. This allowed FDA a fresh overview on PFAS' use in cosmetics, which inspired recent research.
🇺🇸 Research: A December 2025 report revealed that 51 types of PFAS are intentionally used in 1,744 cosmetic formulations in the US, commonly in makeup and even baby products.
🇺🇸 Conclusion: Due to a lack of critical toxicological data and acute toxicity, the safety of 76% of these compounds could not be definitively established. FDA deemed current evidence insufficient to justify a federal ban, opting instead for continued monitoring.
🇺🇸 Note: The FDA excluded environmental considerations and the assessment of unintentional degradation products, which are often the most harmful (e.g., PFOA and PFOS).
EU precautionary principle
🇪🇺 Context: The EU is already phasing out PFAS over concerns regarding long-term health effects and environmental contamination.
🇪🇺 Research: Rising concentrations in water streams and human blood (even in teenagers) are increasingly suspected to suppress the immune system and increase risks of cancer, infertility, thyroid dysfunction, and metabolic dysregulation.
🇪🇺 Conclusion: Action and monitoring stepped up at national and union level.
> This month, France has banned PFAS in all cosmetics (as well as clothing textiles and ski waxes).
> Yesterday, the European Environment Agency (EEA) kicked off a mandatory EU-wide program to systematically monitor PFAS in drinking water.
> Meanwhile, European Chemicals Agency (ECHA) is evaluating a proposal to ban 10,000 PFAS as a broad category, with stricter concentration limits (ppb levels) expected by October 2026.
🇪🇺 Note: The EU had already restricted all PFAS and even banned some under the REACh and the POPs regulations (which also impact allowed limits in medical devices under MDR).
Which side would you take? Personally, I’m leaning EU on this one.
Sources:
- FDA’s report
- EEA programme
- Forever pollution project (image credits)

FDA’s new guidance on general wellness
This post critiques the widening regulatory gap between the US and EU following the FDA's new wellness guidance, highlighting how lower barriers for bold health claims in the US may sacrifice essential quality drivers and complicate global strategies for startups.
Yesterday's release by FDA on wellness vs medical device leaves me with a bitter aftertaste. Why?
I'm usually enthusiastic about policies that lower the barrier to market entry for health products. I'm less enthusiastic about those that eliminate the quality drivers from it..
My main concerns under this guidance:
> General wellness products have no QMS requirement, especially digital ones. So when the guidance says you can now display biomarkers even with some disease reference as long as "the product has validated values" for those biomarkers, it doesn't really mean anything. How do they validate? According to what? Where? Claims get bolder and accountability weaker.
> We will see more products being Class IIa medical devices in EU (with QMS auditing and device file review) while facing zero expectations in the US as general wellness.
> The gap between EU and US regulatory approach gets wider. EU released a "similar" guidance in Sep 2025 emphasising the opposite, with increased focus on mechanism of action and technology rather than relying on claims only. US heads the other way, making it all the more complicated for us RA 🥴
> It will be harder for startups to design their product and strategies for the two main western markets simultaneously. They will be pushed even heavier towards wellness-first but in my experience they get easily stuck there.
> This bold approach may be (too) specific of this administration. Will it then outlive it? It is also clearly result from the WHOOP controversy, given the number of references to Blood Pressure measuring wrist-worn devices. Pretty solid legal and lobby teams there.
One example that puzzles me in particular is the one about glucose monitoring via "minimally invasive microneedle technology" for which FDA says they will apply enforcement discretion as a low risk device. Since I'm currently working on the biocompatibility testing requirements for a device that is hand held by doctors using gloves (👀), I cannot help but finding it unfair towards the rest of the sector.
So I hope you will excuse my slightly less upbeat post this time.
I'm generally excited about the expansion of the definition and agree with the rationale of most of the examples provided.
I'm curious to see what it will mean for international harmonisation and for the opportunities it will open for my clients at this interface!

PCCP beyond AI
Very exciting trend of femtech apps integrating with wearable data! How does this work for the regulated ones? I wanted to share this clever use of PCCP from Natural Cycles° from last year which impressed me.
What's PCCP?
Pre-determined Change Control Plan is a regulatory instrument devised by FDA - as a European is I'm most jealous of. It was designed to enable AI devices, which by design need to be able to evolve their accuracy in the field, getting smarter the more data they acquire. Traditionally, any change to the accuracy and performance of a device required a regulatory resubmission (still the case in EU) and up to 90 days of review wait.
With PCCP you can get pre-approval for a reasonable range of performance that you anticipate and accept.
What I found clever, is that Natural Cycles°, the pioneer of regulated fertility awareness, used PCCP not for AI changes but for variability of source data from different wearables.
While, as far as I'm aware, they currently integrate only with ŌURA and Apple Watch, this clears the way for them to swiftly add any more integrations to their conception/contraception suite as long as they fit their predefined specs (see table in pdf).
This is an example of how:
1️⃣ Regulatory instruments that are smart and abreast with the times enable even more innovation than what they primarily intended to,
2️⃣ Femtech is riding the wave of biomarkers ensuring most users can be served irrespective of which devices they choose - it's not just the iOS vs Android divide anymore!
3️⃣ Scientific research and clinical partnerships will see an incredible boost of opportunity from all this data, finally compensating for the lack of data that we know womens health has suffered until now!
What else could we use PCCP for? And until when can we have a similar toolkit in Europe under MDR? 🫠
NC's current integrations here
Link to full 510k summary here

Review timelines for FDA 510k clearance
How long does it take from FDA submission to clearance?
Let's look at the recent data.
The 510k database can be exported and analysed. Format is not humanly readable but makes a fun ChatGPT exercise.
Here is the result of me playing with the database from devices cleared last months (Aug and Sep 2025).
❗ The normal distribution appears to peak around 90 days, the legal obligation for FDA to respond to submissions. Around 30% of submissions were cleared within that timeframe.
❗ Nice peak at 30 days - but don't be too wishful! These are expedited reviews, e.g. changes to existing 510ks or based on prior agreements or expected updates.
❗ Less exciting peak around 270 days, i.e. 9 months. Most submissions receive an Additional Information request, which gives manufacturers 180 days to respond and restarts the clock for FDA after that (further 90 days).
Lesson here?
If you're planning a 510k, a realistic estimate for clearance is nothing less than 6 months. This is what applied to 2/3s of the 400+ applications cleared most recently.
Good quality submissions and preliminary discussions with FDA on the fundamental topics can help prevent Additional Information requests and thus increase the chances of receiving clearance within 90 days.
Does your experience confirm this too?
I will dig more into this database in the coming posts with more insights.

US Gov shutdown: impact on FDA operations
After Republican and Democratic politicians could not agree to pass a bill funding government services, on 1st October the US federal government has shut down. Though not unusual (almost every administration had at least one, lasting from a couple of days to a top 35 days), they create immediate uncertainty for largely Congress-funded agencies such as the FDA.
FDA announced that, based on its contingency plan, it will limit its ops to “mission critical activities including responding to public health emergencies, supporting high-risk food and medical product recalls, and conducting essential surveillance of medical devices and other medical products”.
So in practice, until the end of the shutdown:
🔴 No new submissions accepted (510k, DeNovo) nor payments thereof,
🟡 Ongoing reviews will continue but may suffer delays beyond the mandatory timeframes and potential unresponsiveness,
🔴 Annual fees will not be processed (MDUFA user registration), though due in October for Fiscal Year 2026 - see my previous post on increased fees,
🟢 Medical device recalls and safety surveillance will continue,
🟡 Inspections largely on hold except if “for cause”.
Tough news if you are on the brink of submitting or awaiting a decision. But history tells us these don't last long, so be ready to move fast once the shutdown lifts.

The WHOOP saga
WHOOP ’s current FDA row is properly binge-worthy. Material for the next Lincoln Lawyer season on Netflix?
But until then, some personal reflections on why it matters for digital health and wearables.
This season’s hottest episodes:
🎞️ Ep. 1 : WHOOP launches Blood Pressure Insights (BPI) as a Wellness feature but claiming medical grade insights.
🎞️ Ep. 2 : FDA’s surveillance picks it up and issues a Warning Letter (made public with exceptional urgency) arguing against the medical disclaimers given the “inherent association” of BP with the diagnosis of hypo/hypertension,
🎞️ Ep. 3 : WHOOP refuses to pull the feature and takes it public/political, meeting with RFK Jr and attacking FDA’s integrity on social media.
I get it, it’s tough to live on the line. Enjoying the aura of “medical-grade” without the burden is the dream of many, but it's getting harder. I’ve been there with multiple startups, and deeply empathise with some of the operational and financial challenges they faced in getting that balance right - often in absence of clear guidelines.
But now: guidance is there, WHOOP already has an FDA-cleared ECG feature (i.e. a QMS) and likely the budget... then why not route the BPI feature under their existing regulated org? Whether from the start or in response to the warning. How is taking up this massive fight a better strategy?
In smaller cases, it would ring a quality culture and integration issue. But in this one, it’s seems a fight on principle - while enjoying the extra PR of being the torch bearer for the freedom of wearables worldwide.
Meanwhile, Hilo by Aktiia quietly secures BP clearance with medical indication for its bracelet without the fuss. 👀
If you’re in the borderline medical space, this is a defining moment.
➡️ Disclaimers may be shorter-lived than ever, careful if you’re relying on those.
➡️ Not all companies are the WHOOP or SPACEX of the ton. Don’t assume this aggressive strategy would work for you, play smart yes, but sustainable. PR and legal repercussions can be devastating for fundability.
➡️ Hire QARA professionals who know how to navigate the redlines vs the negotiables of borderline products.
As Blythe Karow put it in her BEAUTIFUL long read on this story:
“The art lies in reading between the lines and addressing the specific compliance issues rather than fighting fundamental regulatory doctrine.”
Meanwhile, in real life: A friend told me "my sleep/stress score from my watch is looking weird... am I sick??". Familiar? Apparently, WHOOP had an internal policy in place during COVID that employees should stay home if their score was lower than a certain threshold - they either had the virus or could easily get it. If this is how we use these tools, what's so bad in providing assurance of quality and accuracy in the first place?
Only time will tell.. For now, pass the popcorn 🍿

Steep rise in FDA fees for 2025-2026
Alert 🫰 Steep rise in FDA fees from this October:
+19% Annual establishment registration fee from $9,280 to $11,423 (this is the one you pay every year for keeping the right to place a device on the market)
+7% Application fees, e.g. 510k submission from $24,335 to $26,067 (this is the one-off fee for review of a product submission file)
Bad news for early stage medtech businesses and SMEs, in particular since no "small business discount" nor waivers apply on the establishment fee at first registration.
Note, small businesses may qualify for waiver on the establishment fee (2nd year on) and a reduced application fee (e.g. 510k for $6,517 instead of $26,067, new fees) under the SBD programme. Conditions are based on gross sales and justification of "financial hardship", rather than on company size. Worth looking into.
See latest MDUFA fees on the FDA website at this link.

Quality whistleblower - hero vs martyr
How do you make yourself heard when you MUST raise the redflag over design quality, production compliance, clinical safety?
It's an incredibly difficult position to be in, whether you're acting from inside a company or as an external reviewer, stakes are high and office politics (if not even higher politics), budget concerns, along with own self-limiting beliefs, come into play giving you many reasons why you shouldn't follow your gut. Maybe I'm wrong, maybe it's all well. Or maybe it isn't?
I've been in this position before a couple of times as PRRC. It's dire, sleepless nights, conflict escalation. Escalate it to whom? If the technicians or QA's voice is not heard, and your voice as PRRC is not heard, then you hope external parties such as lawyers, consultants, CROs, reviewers will be more effective gate keepers, but then they aren't. They may overlook things or also have their own interests at play. Then who is left to protect the patient? Who is going to stand up and stop the chain of events before it's too late?
The story of Frances Oldham Kelsey, FDA medical reviewer in the 60s who refused to approve Thalidomide is a great example, and similarities can be seen in other preventable disasters such as Titan's OceanGate, Boeing's 737max MCAS software, or Chernobyl to name the most famous. All had a long chain of brave flag raisers in a culture that shut them down..
Culture is key and of utmost importance in medtech. Accountability, feedback and psychological safety create space for risks to be raised and taken seriously at any stage of a project. So called "Type 1 decisions" in business, i.e. non-reversable (launch or not launch?) need true raw information, not just the glossed version that the manager is willing to lend an ear to.
A culture that integrates Quality as their biggest asset and strategic partner will value anyone who raises issues, mistakes, inefficiencies, with a view of preventing not only harm but also resources and reputational risks.
I'm so deeply passionate about driving such cultural shifts and help teams innovate in the most progressive, forward-looking and responsible ways.