

The risk in having too many risks…
Confusing failure modes for risks is one of the most common structural mistakes in medical device risk analysis — and one of the most costly to fix later. This article explains the difference between hazards, hazardous situations, and harm under ISO 14971, why a bloated risk analysis undermines your whole risk management process, and how one simple syntax rule can help you build a cleaner, more actionable document from the start.
Do you have more than 40 risks in your device risk analysis — and it's not even invasive?
Most likely, they are not risks. They are failure modes. And confusing the two is one of the most common — and costly — mistakes I see in early-stage medtech.
A risk list that has grown out of control creates real problems:
It dilutes focus away from the risks that actually matter — the ones you should be able to recite off the top of your head. It opens the door to inconsistencies and duplication in a document so large that no colleague will review it in detail, but that an auditor will flag immediately. It turns every product feature into a hazard or risk control, which then warrants stricter testing requirements down the line. And it makes traceability in post-market surveillance and clinical evaluation a genuine operational nightmare.
I've seen many well-meaning startups suffer through the consequences of a badly designed risk analysis. The QARA who built it might feel proud of its thoroughness. But the rest of the team loses interest and never truly owns their risk areas. Management stops using it for decision-making. Product design becomes cluttered with risk controls — warnings, untouchable features — that nobody can explain.
This kills the collaborative and iterative spirit that is essential for good risk management.
So what's the difference between a risk and a failure mode?
A risk analysis table is built from three distinct layers, as described in ISO 14971 and ISO 24971:
Hazard categories — the nature of the potential harm (energy, software, misuse — full list in ISO 24971)
Hazardous situations — the circumstances in which people are exposed to a hazard, including failure modes and external causes
Harm — the actual injury or damage to health that may result
The most common mistake is conflating hazards with hazardous situations — that is, treating failure modes as if they were risks in their own right. The terminology doesn't help, admittedly.
One simple strategy to keep your risk analysis clean
Use a fixed syntax to write your risks consistently. Here's one I find practical:
THERE IS A RISK OF [who] [hazard type faced] ORIGINATING FROM [list of failures and hazardous situations] WHICH MAY LEAD TO [harm type — pick only the highest level]
Two examples:
For a hardware device: There is a risk of the patient coming into contact with high voltage (electrical energy), originating from a) damage to the connecting cable, b) manufacturing defect, c) poorly designed insulation — which may lead to electric shock.
For a SaMD: There is a risk of the physician receiving inaccurate output from the device (incorrect medical decision), originating from a) algorithm design limitations, b) algorithm execution error, c) user interface failure, d) cybersecurity attack, e) unclear instructions for use — which may lead to delay in treatment.
Notice how multiple failure modes collapse into a single, well-defined risk. That's the point. Your risk analysis becomes shorter, more focused, and far easier to maintain over time.
If you're building the table manually, write the syntax in your header row. If you're using AI-assisted tools, enter it as a prompt constraint or use it to validate the output. If you're reviewing an existing table, run each row against it.
A risk analysis should be accessible to the whole team, actionable in decision-making, and sustainable as the product evolves. Getting the structure right from the start is one of the highest-leverage things a QARA can do in an early-stage company.
Deep Dive: Getting the Structure Right
Risk Analysis vs FMEA
Both are part of the Risk Management process under ISO 14971, but they serve different purposes and are not interchangeable.
Risk Analysis is mandatory. It is the top-level document that captures your device's safety profile — the full picture of what could go wrong, for whom, and with what consequences. Think of it as the billboard for your device's safety. It needs to tell a meaningful story, not overwhelm the reader with noise.
FMEA (Failure Mode and Effects Analysis) is a supporting analytical method — good practice, and often expected by auditors, but not explicitly required by ISO 14971 as a named technique. It is the drill-down tool: you take each component, subsystem, or process and ask systematically, how could this fail, and what would the effect be?
The same FMEA logic appears under different names depending on the domain:
In SaMD, it is often formalised as a Software Hazard Analysis (required under IEC 62304 as part of software risk management)
In usability engineering, it underpins the Use-Related Risk Analysis (URRA), which traces use errors and abnormal use to potential harm — a core deliverable under IEC 62366-1
In cybersecurity, it is effectively a vulnerability analysis or threat modelling exercise (with reference to MDCG 2019-16 and IMDRF guidance on cybersecurity)
Each of these domain-specific analyses follows the same logic: identify how something could fail, then trace that failure to a potential harm. The outputs of all of them feed into one Risk Analysis for your product — not multiple separate risk documents.
This is where the structural confusion often starts. Teams run an FMEA, a URRA, and a software hazard analysis, and then copy the failure modes directly into the Risk Analysis table. The result is a document that mixes hazards, hazardous situations, and failure modes in the same column, under the label "risk." Multiply that across a product with many subsystems, and you quickly reach 60, 80, or 100+ rows — most of which are not risks at all.
The three-layer structure
ISO 14971 and its companion standard ISO 24971 are clear on the terminology, even if teams frequently blur the distinctions in practice:
Hazard: a potential source of harm — an inherent property of the device or its environment (e.g. electrical energy, ionising radiation, software decision output)
Hazardous situation: the circumstance in which a person is exposed to a hazard — this is where failure modes, use errors, and external conditions live
Harm: the physical injury or damage to health or property that results
A well-structured risk analysis row moves through all three layers. The failure modes — however many there are — belong in the hazardous situation column, not in a row of their own. That single structural choice is what keeps the document manageable.
A note on harm classification
For the harm column, the IMDRF Adverse Event Terminology provides a standardised, hierarchical coding system that is increasingly expected in technical documentation and is directly useful in post-market surveillance reporting. Using it consistently from the start — rather than free-text descriptions — saves significant effort later when feeding into your PMSR or PSUR.
Practical checklist
Can every row in your table be read using the [who / hazard type / originating from / harm] syntax? If not, it may be a failure mode, not a risk.
Are failure modes consolidated under their parent risk, rather than listed as standalone rows?
Is the harm column using consistent, ideally IMDRF-aligned terminology?
Could a new team member read the Risk Analysis and understand the device's core safety story in under 30 minutes?
References
ISO 14971:2019 and ISO 24971:2022 — available at a significantly lower cost than ISO directly via the Estonian Standards store (legitimate national standards body, same official text)
IMDRF Adverse Event Terminology browser — for standardised harm classification
IEC 62304 (software lifecycle) and IEC 62366-1 (usability engineering) — for domain-specific hazard analysis requirements that feed into the Risk Analysis
Methodology note: This article is based on two original LinkedIn posts (first, second) written by me, reflecting my professional experience and personal perspectives on risk management in medical device development. Claude AI assisted in combining and expanding the posts into a broader article for this blog, integrating background context, regulatory references, and a structured Deep Dive section. All regulatory perspectives and practical recommendations are my own, and all content has been reviewed by me for accuracy.
The next strategic frontiers of medtech
Where should medtech and femtech companies focus their international expansion efforts after establishing EU and US market presence? This article analyses eight high-potential markets across four global regions — Anglosphere, East Asia, MENA, and Latin America — ranked against three strategic criteria: ROI and medtech infrastructure, femtech readiness, and regulatory alignment with international harmonisation frameworks including MDSAP and CE Mark mutual recognition agreements. Whether you are mapping your next regulatory strategy or building a market entry roadmap, this evidence-based analysis offers a practical starting point — with an honest look at what the data says and where gut instinct still has a role to play.
After my popular recent poll, which strategic markets deserve to be your and my next medtech focus beyond US-EU? Note: beyond, not instead of!
What are the factors we want to optimise?
ROI & MEDTECH INFRASTRUCTURE: markets that offer the highest investment and monetization potential, with advanced payer structures and tech scaling capabilities.
FEMTECH READINESS: regions where the cultural and policy discourse is opening to women’s health, where clinical research is advanced, and femtech innovation and startup ecosystem are thriving.
REGULATORY ALIGNMENT: countries that align with international medtech harmonisation efforts, for example being members of MDSAP or with mutual recognition agreements (MRA) towards CE mark and FDA.
With the help of Claude, I ran an advanced analysis to find the right countries for these categories. More details on the results summarised below.
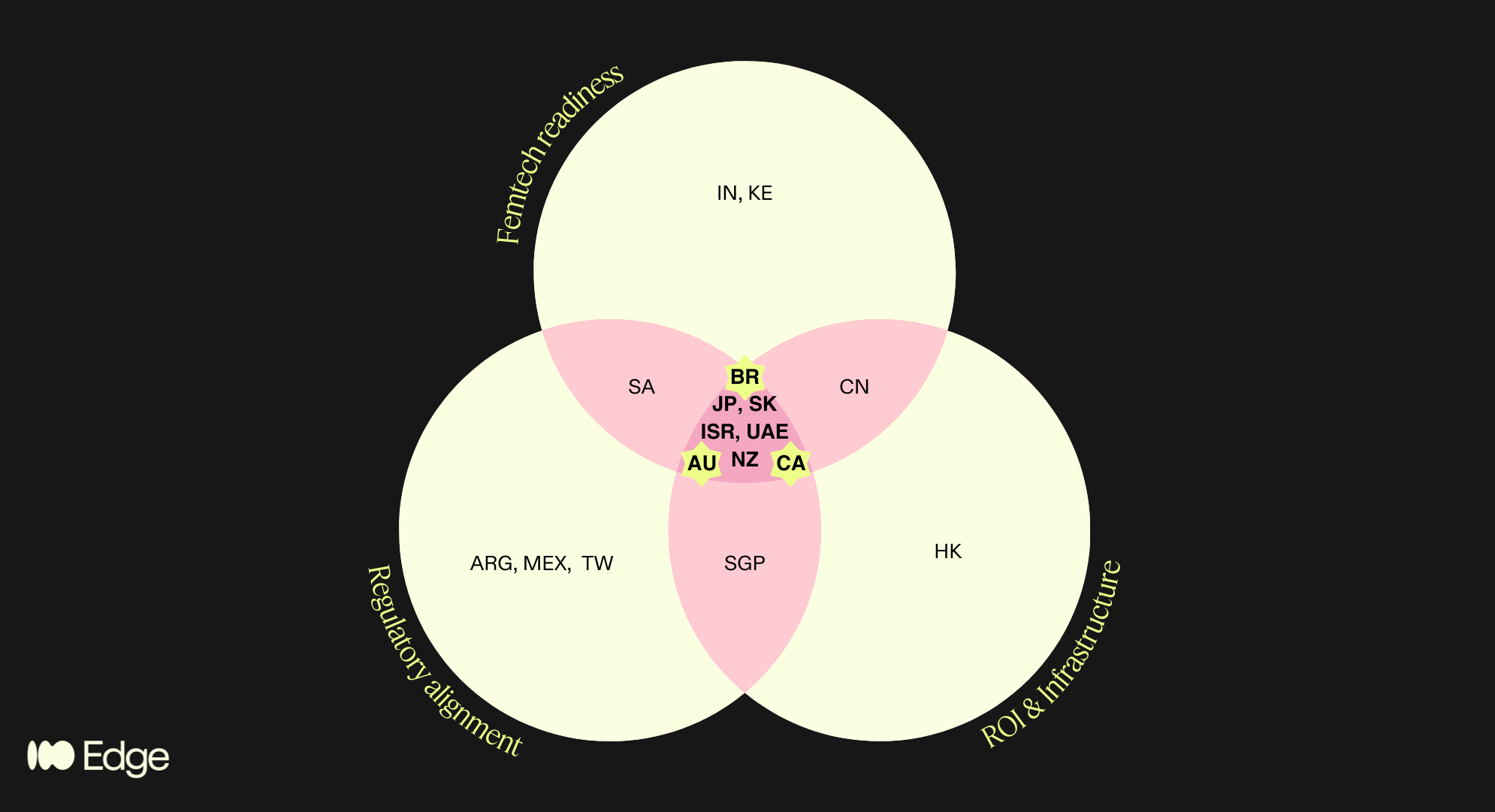
Regions that satisfied multiple criteria are highlighted in the pink areas of the diagram. This brings us to 8 high potential regions, spanning the 4 geographies of my poll!
Anglosphere: Canada, Australia, New Zealand
“BRIC”+East Asia: Japan, South Korea
MENA: Israel, UAE
LatAm: Brazil
What does it mean for your market strategy? If you are in femtech and looking to expand after EU/US proof-of-concept - AND not planning to exit directly - then these are your markets. Save and share the post, and narrow down the list based on further criteria relevant to you and your business.
To me and my business, I applied the following additional criteria:
Accessibility: where can my cultural roots and the languages I speak help me?
Market size: >20M for highest chances of leads and indirect product reach for my clients
Heterogeneity: the more diverse the better for clinical research relevance
Geopolitical stability: low risk of conflicts and market volatility
Which takes us to the winners!
🇧🇷 Brasil: as half-Brazilian I’ll be proud and excited to explore the femtech, clinical and regulatory space and build connections there! DM me to discuss!
🇦🇺 🇨🇦 Australia and Canada: it’s a tie! I already work with these markets, no big lift, but good pointer to go a bit deeper. Spoiler: more MDSAP content coming, like it or not..!
🇦🇪 UAE: Although it didn’t pass my filters, something in my gut tells me not to give up on it. I’d love to partner up with medtech experts and providers in that area.
This aligns well with the results from the poll: 52 votes, indicating predominant interest in "anglosphere" (32%) and "emerging" (30%), incl. LatAm.
Thanks to all who voted, commented and even called me to share their passionate insights!
Deep dive
The framework: three lenses, not one
We structured the analysis around three criteria, chosen because they reflect the real bottlenecks that founders face when entering new markets.
Regulatory alignment (MDSAP / MRAs). For a CE-marked or FDA-cleared device, the most efficient path into a new market is one that recognises or aligns with those approvals. The Medical Device Single Audit Program (MDSAP) is the primary international harmonisation vehicle, currently comprising the US (FDA), Canada (Health Canada), Brazil (ANVISA), Australia (TGA), and Japan (PMDA). Beyond MDSAP, a growing network of Mutual Recognition Agreements (MRAs) extends this regulatory compatibility further — most notably, the EU–Australia MRA, Israel's deep alignment with EU MDR, South Korea's MFDS harmonisation pathway, and GCC's increasing adoption of CE and FDA as reference standards.
Femtech readiness. This goes beyond market size. It asks whether the cultural, policy, and clinical infrastructure of a market is genuinely open to women's health innovation — or whether femtech companies will spend the first two years of market entry fighting the problem rather than solving it. We looked at startup density, clinical research activity, regulatory openness to digital health, and the status of women's health policy discourse in each candidate country.
ROI and medtech infrastructure. Commercial viability ultimately depends on whether a market can pay for solutions and scale them. This means evaluating healthcare financing structures (private payer vs. public reimbursement vs. out-of-pocket), digital health infrastructure, startup ecosystem depth, and the presence of credible distribution and partnership networks.
Only markets that scored meaningfully across all three lenses made the final shortlist. That intersection — regulatory alignment AND femtech readiness AND commercial infrastructure — is the filter that separated 8 markets from the 25+ we considered.
The 25-country universe and how it narrowed
We mapped approximately 25 countries across the three criteria, drawing on data from:
Institute for Economics & Peace Global Peace Index 2024 (163 countries) for geopolitical stability scoring
EF English Proficiency Index 2024 (116 countries) for accessibility
UN Population data 2024 for market size
World Bank ethnic fractionalization data for demographic heterogeneity (relevant to clinical research representativeness)
Vestbee, Dealroom, Speedinvest, Grand View Research, Startups Magazine for femtech market data by country
MDSAP member country documentation and publicly available MRA registers for regulatory alignment
Countries in only one or two circles of the Venn include genuinely interesting markets — Singapore (strong MDSAP alignment and infrastructure but too small for standalone commercial focus), India (exceptional femtech growth projected at 17.8% CAGR to 2030 per Grand View Research, but regulatory alignment is a work in progress), South Africa (emerging femtech ecosystem with strong policy momentum, but infrastructure gaps and geopolitical risk), China (significant commercial potential but regulatory and geopolitical friction makes it a poor fit for EU-anchored companies), and Kenya (one of Africa's most active femtech ecosystems, but not yet operationally viable for most European companies without a dedicated local strategy).
These are not dismissed markets — they are markets for a different risk appetite or a later stage.
The 8 that made the intersection
These are the markets that sat inside all three criteria simultaneously. For each, we break down the case across the three categories.
🇦🇺 Australia
Regulatory Alignment
MDSAP full member (TGA)
EU–Australia Mutual Recognition Agreement in place — CE mark directly supports TGA approval pathway
Strong alignment with ISO 13485 and IEC 62304 for software-based devices
Femtech Readiness
Mature digital health market with active government investment (Australian Digital Health Agency)
Women's health policy discourse is open and progressive; no significant cultural friction for femtech categories
Active medtech and healthtech startup ecosystem, particularly in Melbourne and Sydney
ROI & Infrastructure
Population ~27 million; 27% foreign-born (one of OECD's highest) — valuable for diverse clinical evidence
Mixed public/private payer system; growing private digital health reimbursement
English-speaking, Commonwealth-aligned, low operational complexity
GPI 2024: #17 — high geopolitical stability
🇨🇦 Canada
Regulatory Alignment
MDSAP founding member (Health Canada)
A single MDSAP audit covers Canada alongside the US, Brazil, Australia, and Japan — significant efficiency gain
Health Canada increasingly aligned with FDA on digital health device classification
Femtech Readiness
Toronto and Vancouver are active femtech hubs; Femtech Canada is an established national ecosystem
Strong clinical research infrastructure with diverse patient populations
Progressive women's health policy environment; high awareness and destigmatisation of femtech categories
ROI & Infrastructure
Population ~40 million; ~27% immigrant share — comparable diversity to Australia
Robust private and public payer landscape; provincial variation requires navigation
English and French markets; English operations are broadly sufficient for market entry
GPI 2024: #13 — highly stable
🇧🇷 Brazil
Regulatory Alignment
MDSAP full member via ANVISA — the most underutilised structural advantage for European companies entering LatAm
A single quality audit unlocks market access across all five MDSAP jurisdictions simultaneously
ANVISA has been modernising its device framework; registration timelines have improved significantly
Femtech Readiness
Growing digital health and femtech ecosystem, particularly in São Paulo and Rio de Janeiro
Large, young, highly digitally engaged female population
Significant unmet need in reproductive health, maternal health, and menstrual care — high receptivity to femtech solutions
ROI & Infrastructure
Population ~215 million — the largest in Latin America by a substantial margin
Highest ethnic fractionalization score globally (World Bank) — clinical data generated here is maximally representative
⚠️ GPI 2024: #131 — operational complexity is real; security considerations, contract risk, and local partner dependency must be factored in
Local partner infrastructure is a prerequisite, not an option
🇯🇵 Japan
Regulatory Alignment
MDSAP full member via PMDA (Pharmaceuticals and Medical Devices Agency)
PMDA is one of the most rigorous regulatory bodies globally; MDSAP audit significantly reduces duplicative burden
Regulatory pathway for digital health devices (SaMD) is well-defined
Femtech Readiness
One of the world's most advanced femtech markets by per-capita spend on menstrual health and reproductive technology
Strong clinical research infrastructure and data quality
⚠️ Cultural norms around women's health are evolving but remain conservative in some categories — market education investment required
ROI & Infrastructure
Population ~124 million; high healthcare spending and premium consumer health culture
High-margin market for validated, evidence-backed products
⚠️ EF English Proficiency Index: #92 out of 116 — significant language barrier; local partner or Japanese-speaking regulatory lead is non-negotiable
GPI 2024: #11 — very high stability
🇰🇷 South Korea
Regulatory Alignment
MFDS (Ministry of Food and Drug Safety) has progressively aligned with FDA and EU MDR frameworks
MDSAP compatibility pathway is expanding; not yet full membership but convergence is directional
Government actively promoting K-healthcare internationally — creating reciprocal openings for foreign market entry
Femtech Readiness
High digital health adoption and smartphone penetration; strong consumer appetite for health tracking and wearables
Growing awareness of women's health, particularly in fertility and menstrual health tech
Active startup ecosystem with increasing women's health focus
ROI & Infrastructure
Population ~52 million; advanced payer structures and tech scaling infrastructure
Government investment in digital health innovation is substantial
⚠️ EF EPI: #50 — business English limited outside Seoul; local presence required
GPI 2024: #46 — stable, with standard geopolitical considerations given peninsula context
🇮🇱 Israel
Regulatory Alignment
Israeli MOH operates with strong alignment to EU MDR and FDA standards — one of the most harmonised non-MDSAP markets globally
CE mark is a recognised reference standard; dual EU/FDA submission strategies are well-established
Deep bilateral ties with both EU and US regulatory bodies
Femtech Readiness
Exceptionally strong clinical research infrastructure; Israel produces a disproportionate number of medtech innovations relative to population
Highly developed VC and medtech ecosystem; femtech companies including HeraMed are globally recognised
High awareness and openness to women's health innovation; destigmatisation is advanced relative to regional peers
ROI & Infrastructure
Population ~10 million — small absolute market, but high income and high health spend per capita
English proficiency high (EF EPI: #51); business environment is highly accessible to European operators
Strong potential as a clinical trial site and R&D partnership hub, regardless of commercial scale
⚠️ GPI 2024: #155 — reflects active conflict situation as of the 2024 index; market entry decisions require live geopolitical risk assessment, not a static one. The underlying regulatory, clinical, and commercial infrastructure remains among the strongest in this cohort
🇦🇪 GCC (UAE & Saudi Arabia)
Regulatory Alignment
Neither is a formal MDSAP member, but UAE and KSA have adopted CE mark and FDA clearance as primary reference pathways for device registration
UAE MOHAP and Saudi SFDA increasingly streamlined; CE mark provides a strong starting point
GCC Standardization Organization (GSO) harmonisation creates some regional regulatory efficiency
Femtech Readiness
Saudi Arabia's Vision 2030 explicitly references women's empowerment and healthcare improvement — femtech is policy-aligned
UAE has the most open and internationally diverse women's health market in the region
Growing investor interest in femtech and digital health across the Gulf; GITEX Health and Arab Health are active deal-making forums
ROI & Infrastructure
UAE: ~89% of resident population is expatriate (UN data) — genuinely diverse patient population and cosmopolitan business environment
English is the functional operating language for business in both markets
Premium private-pay market; reimbursement structures favour high-value, evidence-backed products
⚠️ GPI 2024: UAE #53 (improved 31 places in 2024 — single largest improvement in the index); KSA requires ongoing monitoring
Cultural distance to women's health topics exists in some categories — local partnership and community navigation are important
🇳🇿 New Zealand
Regulatory Alignment
MDSAP full member (Medsafe)
Strong regulatory alignment with Australia (TGA); joint Trans-Tasman regulatory considerations apply
Simple, transparent regulatory environment — among the most accessible globally for device registration
Femtech Readiness
Progressive women's health policy environment; high health literacy and digital engagement
Maori and Pacific women's health is an active policy and research priority — relevant for inclusive femtech design and clinical evidence
English-speaking, low cultural friction across all standard femtech categories
ROI & Infrastructure
Population ~5 million — too small for standalone commercial focus
Best positioned as a clinical trial jurisdiction, regulatory proof-of-concept market, or Australasian stepping stone alongside Australia
GPI 2024: #4 — among the most geopolitically stable countries on earth
High income per capita; public system (Pharmac) for reimbursement is conservative but private market is accessible
What this means for women’s health startups
The eight markets are not interchangeable, and we are not suggesting a company should pursue all of them simultaneously. The practical read is this:
For a femtech company at the EU/US proof-of-concept stage, with limited expansion resources and no existing presence outside Europe, Australia and Canada are the lowest-friction first moves. They are MDSAP members, English-speaking, culturally accessible, and have enough market depth to justify the investment.
Brazil becomes compelling the moment you have a local partner or are prepared to build one. The MDSAP pathway is a structural advantage that is consistently underutilised by European companies, and a population of 215 million — with the highest demographic diversity of any market on this list — is a significant clinical research and commercial prize.
Japan and South Korea are high-reward markets that require deliberate localisation investment. The regulatory frameworks are accessible through MDSAP and MRA alignment; the human infrastructure needs to be built.
Israel and the GCC are specialist considerations — Israel for companies with strong clinical trial and research ambitions, the GCC for companies targeting the premium private-pay women's health segment and building into the Middle East and North Africa corridor.
New Zealand is genuinely valuable as a regulatory and clinical site — less so as a primary commercial target.
The 4th lens…
At Edge Compliance we want to keep serving US and EU market entries (EU as a continent, incl. UK and CH).
What this analysis taught us is where to put our next focus in terms of partnerships, network, learning, advocacy.
In order to avoid spreading ourselves too thinly, we applied some filters to narrow down the list of high potential regions to a smaller selection. The Edge Compliance lens considers:
Cultural accessibility
Market size
Demographic heterogeneity
Geopolitical stability
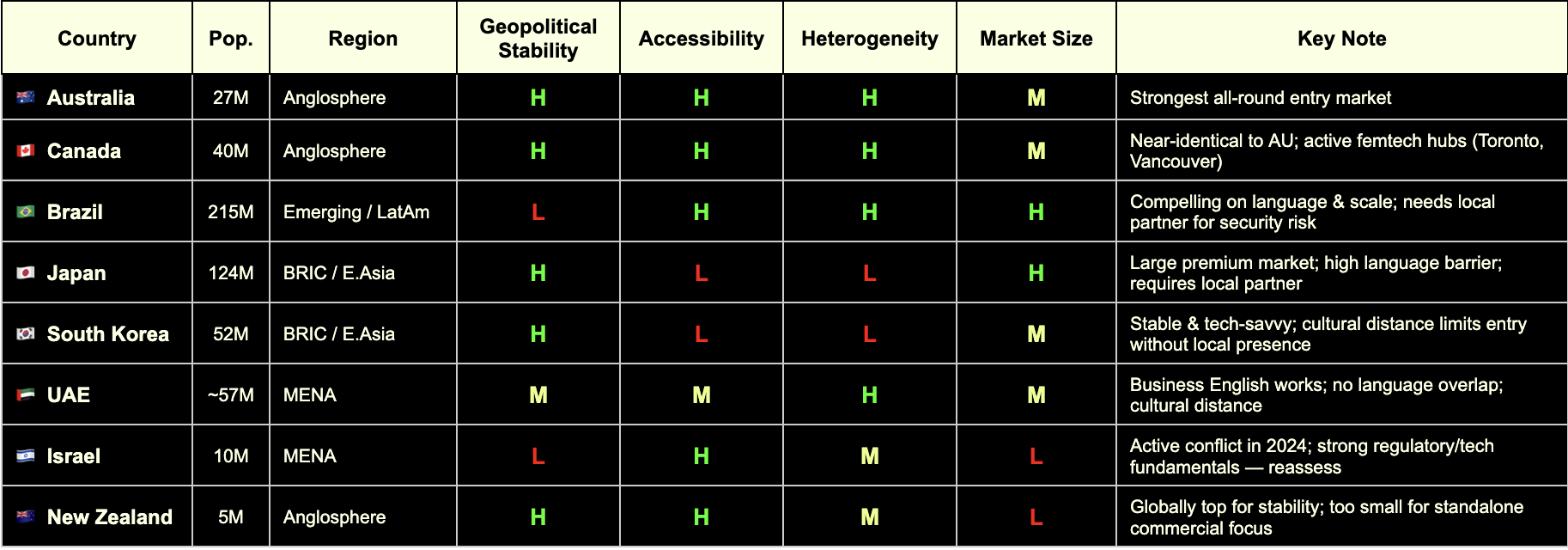
Criteria for market selection for Edge Compliance. Which are relevant to your business?
This analysis led us to choose the following areas of focus in addition to our existing focus and expertise:
🇧🇷 Brasil
🇦🇺 Australia
🇨🇦 Canada
🇦🇪 UAE
We trust this will help us help YOU with future-proof regulatory strategy and foresight that is ahead of trends.
References
Institute for Economics & Peace GPI 2024
EF English Proficiency Index 2024
UN World Population Prospects 2024
World Bank Ethnic Fractionalization Data
Vestbee Femtech Market Overview (2024)
Dealroom Femtech Report (2024)
Speedinvest Femtech Investment Analysis
Grand View Research India Femtech Market Outlook 2024–2030
MD+DI FemTech Analytics (2024)
Startups Magazine Southeast Asia Femtech Report
The Recursive CEE Femtech Analysis (2025)
Methodology note: This Deep Dive is based on my original LinkedIn post, reflecting my professional experiences and personal perspectives. Claude AI assisted in elaborating the topic into a broader article by integrating personal notes, literature research, fact-checking and deeper insights on the topic. All analysis and regulatory perspectives are my own, and all content has been reviewed by me for accuracy.

Quantum: The Regulatory Frontier That Will Catch Us Off Guard
Quantum computing is revolutionizing medical device development, but regulatory frameworks aren't ready. Companies like Algorithmiq are achieving 100x precision improvements in cancer therapy using quantum-generated synthetic data—yet FDA's 2025 AI/ML guidance doesn't address quantum validation challenges. How do you validate data you can't reproduce classically? Regulatory science needs quantum-aware frameworks now, before quantum-AI medical devices reach the clinic.
Yesterday, I met Sabrina Maniscalco, CEO of Algorithmiq, at the Italian Tech Forum in Zurich. They develop quantum algorithms for life sciences, including applications in cancer therapy. Despite having taken two courses in quantum physics at university and working extensively with AI/ML medical devices, I found myself needing to educate myself from scratch on what quantum computing means for our field.
What I discovered was both inspiring and unsettling: we're standing at the edge of a paradigm shift in medical technology, and regulatory science isn't even looking in that direction yet.
What Is Quantum Computing and Why Does It Matter?
Classical computers operate using bits, each either 0 or 1. Quantum computers use qubits that can exist in multiple states simultaneously through superposition. Qubits can also be entangled, meaning their states are interconnected regardless of distance. This allows quantum computers to process vast possibilities in parallel in ways classical computers simply cannot replicate.
For medtech, this isn't academic curiosity—it's transformative capability. Classical computers struggle to simulate even simple molecules beyond a certain size. A molecule with just 30 atoms has more quantum states than a classical computer can practically track.
Quantum computers can model:
Drug-molecule interactions at atomic precision
Protein folding and three-dimensional structures
Tumor microenvironments at cellular and molecular levels
Personalized treatment responses based on genetic profiles
The AI-Quantum Hybrid Approach
During our conversation, Sabrina explained: "AI is only as good as the data it's trained on. With quantum computing, we can generate vast amounts of physically accurate data and then train AI on it. Imagine the possibilities from simulating the behavior of ALL atoms in our body."
This is Algorithmiq's innovation: combining quantum computing with AI to address one of AI's fundamental limitations—the need for massive training data.
For many biological phenomena, we simply don't have enough experimental data. It's too expensive, too dangerous, or physically impossible to measure. Quantum computers can generate synthetic training datasets that are physically accurate—based on quantum mechanics—but impossible to obtain experimentally.
Sabrina mentioned that quantum computing capabilities are now available on the cloud, with enterprise access in the million-dollar range annually. For specific computational problems, quantum computers can be more efficient than traditional supercomputers.
Algorithmiq has already announced partnerships with Microsoft (December 2024) and Quantum Circuits (February 2025) to accelerate drug discovery.
A Concrete Example: Photodynamic Cancer Therapy
One particularly compelling application demonstrates quantum computing's real-world impact: photodynamic therapy (PDT) for cancer.
PDT uses special molecules called photosensitizers that are activated by light to produce therapeutic effects. The benefits are significant:
No long-term side effects
Less invasive than surgery
Outpatient procedure
Precisely targeted
Can be repeated at the same site (unlike radiation)
5-10 times less costly than other cancer treatments
The challenge lies in designing these photosensitizer molecules. It requires understanding tiny energy gaps between electronic states—differences that dictate how molecules behave when exposed to light. Classical quantum chemistry algorithms struggle to calculate these energy gaps with the necessary accuracy.
Using IQM's Emerald quantum processing unit and Algorithmiq's advanced error mitigation techniques, the team achieved a 100x improvement in precision compared to results from other quantum hardware providers. This work, part of the Wellcome Leap Q4Bio Challenge, is establishing an end-to-end quantum-centric drug discovery pipeline for light-activated anti-cancer drugs.
They're focusing on the BODIPY class of compounds—next-generation photosensitizers. With quantum computing, simulating their energy landscape becomes possible with unprecedented accuracy, paving the way for better-targeted therapies developed faster and more cost-effectively.
This is happening now.
Closing Health Data Gaps
We also discussed possibilities that particularly resonate with my work in femtech: using quantum computing to simulate complex biological systems like women's physiology to close health data gaps that are difficult or impossible to obtain experimentally.
Women's health research has historically been underfunded. Menstrual cycles, pregnancy, menopause—these introduce biological complexity that makes clinical trials more expensive and results harder to interpret. What if quantum simulation could help bridge these gaps by modeling hormonal interactions and reproductive system responses with atomic-level precision?
The Regulatory Challenges Ahead
Here's the uncomfortable truth: none of medtech's regulations or guidances currently contemplate quantum-AI hybrid diagnostics or therapeutics.
Challenge 1: The Validation Paradox
How do you validate quantum-generated data when you can't reproduce it classically?
The FDA's recent draft guidance on AI/ML-enabled device software (January 2025) requires manufacturers to disclose synthetic data provenance, describe algorithms used to generate it, and demonstrate it preserves clinical correlations. These are sensible requirements for classically-generated synthetic data.
But they break down when the "algorithm" is a quantum computer simulating physics that classical systems fundamentally cannot reproduce. How do you verify quantum-generated molecular data "preserves clinical correlations" when there's no classical ground truth? The entire point of quantum computing is simulating phenomena classical computers cannot.
Challenge 2: Black Box Squared
AI is already a "black box"—how do we maintain our ability to explain and reproduce the operating principles when we layer quantum computing on top?
Explainability is already a regulatory challenge. The EU MDR Article 61 and FDA guidance emphasize transparency in clinical decision-making. But AI models, particularly deep learning, are notoriously opaque.
Add quantum computing—inherently probabilistic, extraordinarily sensitive to environmental interference—and we're layering one form of opacity on another. Yet regulatory frameworks require that medical devices be explainable, reproducible, and transparent.
The FDA's three-pillar framework for Software as a Medical Device asks:
Is there a valid clinical association between device output and clinical condition?
Does the software correctly process input data?
Does use of the output achieve the intended purpose?
For quantum-AI systems, how do you analytically validate "correctness" when there's no classical benchmark?
Challenge 3: Cybersecurity and Q-Day
Quantum computers will eventually break current encryption methods—a threat called "Q-Day." This poses serious risks:
Adversaries can collect encrypted medical data today and decrypt it later
Medical devices relying on current cryptographic protocols will be compromised
NIST announced its fifth quantum-safe algorithm in March 2025, but adoption in medical devices has been slow. Medical device manufacturers should implement quantum-resistant encryption immediately.
What Regulatory Frameworks Currently Exist?
The closest we have are two recent FDA guidances:
FDA Guidance on Real-World Evidence (December 2025) emphasizes that data must be relevant and reliable, with a fit-for-purpose approach. This potentially opens a pathway: quantum-generated synthetic data could be acceptable if manufacturers demonstrate it's the most appropriate method for answering specific clinical questions.
FDA Guidance on AI/ML-Enabled Device Software (Draft, January 2025) addresses data management, synthetic data requirements, performance validation—but all assuming classical computational paradigms.
Neither contemplates quantum-generated training data, validation when classical reproduction is impossible, or uncertainty quantification for quantum probabilistic outputs.
The EU AI Act, MDR/IVDR, and ISO standards similarly don't address quantum computing.
Why This Matters Now
Waiting until quantum devices reach the clinic or reach their "ChatGPT moment" means we'll be reactive instead of proactive, again.
We've seen this pattern with AI. By the time ChatGPT brought AI to mainstream awareness, the technology had been developing for decades. Regulators scrambled to catch up.
But AI builds on classical computing principles we already understood. Quantum computing is fundamentally different. The learning curve is steeper, the validation challenges more complex.
If we wait until a quantum-enhanced diagnostic applies for FDA clearance before starting these conversations, we'll be years behind.
So, what needs to happen?
Regulatory agencies may:
Introduce quantum-aware terminology in guidance documents
Establish working groups bringing together quantum scientists, device developers, and regulatory professionals
Develop validation frameworks specifically for quantum-generated synthetic data
Issue guidance on quantum-resistant cybersecurity for medical devices
Industry may:
Engage early with regulators through pre-submission meetings
Document quantum approaches in detail
Build quantum literacy within regulatory and quality teams
Implement post-quantum cryptography now
Thank you to Sabrina Maniscalco for the thought-provoking conversation, and to Camera di Commercio Italiana a Zurigo for creating the space where these insights happen.
References
FDA Software as a Medical Device (SaMD): Clinical Evaluation
FDA guidance: “Use of Real-World Evidence to Support Regulatory Decision-Making for Medical Devices”
FDA guidance: "Software as a Medical Device (SAMD): Clinical Evaluation"
Methodology Note: This article is based on my original LinkedIn post, reflecting my professional experiences and personal perspectives. Claude AI assisted in elaborating the post into a broader article by integrating personal notes, literature research, fact-checking and deeper insights on the topic. All analysis and regulatory perspectives are my own, and all content has been reviewed by me for accuracy.

Beyond the EU-US paradigm
The global medtech regulatory landscape is shifting away from the traditional EU-US duopoly. The UAE now offers approval timelines 30-50% shorter than US and EU markets, while Mexico and Nigeria introduced comprehensive digital health regulations in 2023 and 2025. Canada, Australia, the UK, and Brazil are implementing bold reforms—from the UK's AI Airlock regulatory sandbox to Brazil's platform tracking over 500 registered software medical devices. This analysis explores strategic implications for medtech companies choosing between the Anglosphere, BRIC nations, Middle East markets, and emerging economies for regulatory submissions.
The recent speech delivered by Canada's Prime Minister at the World Economic Forum prompted me to reconsider my approach to regulatory strategy. Without delving into politics, the address highlighted the extent to which the western world has become US-centric and reliant on American frameworks—a reality that extends fully to the medical technology sector.
This led me to reflect on my own professional practice:
Most of my analysis focuses on comparative regulatory matters between the EU and US.
Most of my medtech clients prioritize EU and US market entry first, and that is what I help them achieve.
Most of my knowledge accumulated over 14 years in healthtech has been built on the default EU-US paradigm.
However, I am far from blind to the strong signals of change emerging on a global level.
Major medtech corporations are beginning to pursue UAE market entry first, given that average regulatory approval timelines are 30%-50% shorter than in the US and EU. Many emerging economies maintain low regulatory barriers for standalone Software as a Medical Device (SaMD) in digital health—large markets such as Mexico and Nigeria only introduced such requirements in 2023 and 2025, respectively. Meanwhile, Canada, Australia, and the UK are taking bold steps to boost health innovation, attract technology companies, and facilitate regulatory compliance.
As former Bank of England Governor Mark Carney stated in his remarks:
"In a world of great power rivalry, the countries in between have a choice: to compete with each other for favour or to combine to create a third path with impact."
"The question is not whether to adapt—we must. The question is whether we adapt by simply building higher walls or whether we can do something more ambitious. The former is easy and ruinous; the latter is difficult and necessary."
This brings me to an important question for the medtech regulatory community.
Beyond the US-EU 'old order', which regulatory focus would you find most valuable for future analysis and guidance?
Anglosphere: Canada, South Africa, Australia, New Zealand
"BRIC" + Japan: Brazil, Russia, India, China, and Japan
Middle East: UAE, Saudi Arabia, Israel
Emerging Markets: Africa, ASEAN, Latin America
I invite you to share your perspective in the poll here

Plasters Vs Period Products
Why are tampons regulated differently than plasters? Discover the critical gap between "hygiene products" and Class I Medical Devices (MDR). From ISO 13485 quality standards and biocompatibility testing to the FDA’s Class II classification, we explore why medical-grade manufacturing—like Daye’s cleanroom production—is essential for menstrual safety.
If plasters are Class I medical devices in the EU, why shouldn't tampons be held to the same quality and biocompatibility standards?
It's a question that seems obvious once you think about it. A plaster sits on your skin for a few hours. A tampon is inserted into one of the most absorbent mucosal tissues in the body, often for 4-8 hours at a time, repeatedly, for decades of a person's life.
Yet in the EU, menstrual products—including cups, pads, and tampons—are classified as hygiene products under the General Product Safety Regulation (GPSR), not medical devices. Unless, that is, manufacturers choose to voluntarily market them as medical devices.
Under the Medical Device Regulation (MDR), manufacturers can voluntarily classify their menstrual products as medical devices by applying Rule 4:
Class I, if non-invasive medical devices act as a mechanical barrier for absorption and come into contact with a mucous membrane
Class IIa, if controlling the micro-environment of the mucous membrane or diagnostic
This voluntary pathway exists—but very few manufacturers take it. And here's why that matters.
What's the Difference?
The gap between a hygiene product and a Class I medical device isn't just bureaucratic classification. It's a fundamental difference in accountability and safety standards:
Product Design & Testing
Hygiene product: Basic safety assessments under GPSR
Class I medical device: Thorough documentation and biocompatibility testing for cytotoxicity, irritation, sensitization, and leachables per ISO 10993
Legal Paper Trail
Hygiene product: Non-standardised quality systems
Class I medical device: Mandatory ISO 13485 Quality Management System with full traceability from raw materials to finished product
Production Environment
Hygiene product: "Clean" room (undefined standards)
Class I medical device: Certified cleanroom with documented environmental controls
Surveillance
Hygiene product: Reactive response to complaints
Class I medical device: Proactive post-market surveillance, vigilance reporting, and periodic safety updates
These aren't trivial differences. They're the scaffolding that ensures product safety when something is inserted into your body thousands of times over your lifetime.
US Takes a Different Approach
Here's where it gets interesting: in the United States, menstrual products are regulated as medical devices by the FDA:
Pads: Class I medical devices
Tampons and cups: Class II medical devices (requiring 510(k) premarket notification)
This means, in principle, a higher baseline for quality and safety of menstrual products in the US compared to the EU.
But—and this is important—there are two contrasting factors that complicate this picture:
Factor 1: The Predicate Problem
FDA clearance for Class II devices relies on proving "substantial equivalence" to a pre-existing product (the predicate). For menstrual products, many of these predicates are decades old, with correspondingly low benchmarks.
The 510(k) pathway asks: "Is your product substantially equivalent to this 1980s tampon?" Not: "Does your product meet 2025 safety standards?"
This creates a regulatory floor that hasn't risen substantially in 40+ years. Manufacturers must demonstrate biocompatibility and safety testing, but the comparison point is historical, not state-of-the-art.
Factor 2: EU's Horizontal Chemical Legislation
The EU has stronger horizontal legislation than the US, with general bans on:
Carcinogens, mutagens, and reprotoxic (CMR) substances
Endocrine disruptors
Certain phthalates and parabens
Specific pesticide residues
These bans apply to all consumer products, including hygiene products, through REACH (Registration, Evaluation, Authorisation and Restriction of Chemicals) and the General Product Safety Regulation.
The new GPSR (which fully applied from December 2024) strengthens these protections:
Mandatory risk assessments before market placement
Technical documentation of hazards and mitigation
Designated EU responsible person
Enhanced traceability (batch coding for rapid recalls)
Proactive approach to safety, not just reactive
Additionally, the EU Ecolabel criteria (updated September 2023) ban many hazardous substances from certified products:
Antibacterial agents
Formaldehyde
Parabens
Endocrine disruptors
Phthalates
Allergenic fragrances
So while EU menstrual products aren't medical devices, they benefit from chemical safety requirements that make even hygiene products safer than some US medical devices might be, depending on their predicate.
The voluntary approach: Daye’s case
This is where companies like Daye become important. Founded and led by Valentina Milanova, Daye chose to produce tampons to medical device standards—even though they're not legally required to in the EU.
What does this mean practically?
Cleanroom production: Daye manufactures tampons in an ISO 14644-1 Class 7 cleanroom, the same standard required for medical device production. This isn't just marketing—it's certified, monitored environmental control.
ISO 13485 QMS: Full quality management system compliance, ensuring operations from production to testing to post-market surveillance match medical device requirements. When Daye identifies an issue with a tampon, they can trace every raw material that went into it and check other batches.
Gamma ray sterilization: Medical-grade sterilization to eliminate microbial contamination, reducing TSS (Toxic Shock Syndrome) risk.
Biocompatibility testing: Testing for cytotoxicity, irritation, sensitization—the full ISO 10993 battery that medical devices require.
Batch testing: Testing for pesticides, dioxins, heavy metals, CBD content (for their CBD tampons), THC content.
Full ingredient transparency: Voluntarily disclosing all ingredients and manufacturing processes, even though GPSR doesn't require it.
The result? Products manufactured to the same standard as stents and hip replacements—for something you insert vaginally multiple times per month for 30+ years of your life.
The Daye Warning Letter: A Cautionary Tale
In December 2024, the FDA issued a warning letter to Daye regarding their CBD-infused tampons. The letter highlighted a critical regulatory challenge: Daye's tampons were cleared under K223883 with indications "to absorb menstrual discharge," but their labeling and promotion suggested the device was "intended for relief of period related pain"—a major change in intended use requiring separate clearance.
This illustrates the complexity manufacturers face when trying to innovate in this space. Daye was attempting to address a real medical need (90% of menstruating people experience dysmenorrhea), but doing so triggered different regulatory requirements.
This tension between innovation and regulation is exactly why clear, appropriate frameworks matter.
Why This Matters for Women's Health
The average person who menstruates will use 11,000-17,000 tampons or pads in their lifetime. That's decades of exposure to whatever materials, chemicals, and contaminants are in those products.
Recent research has raised concerns:
A 2024 UC Berkeley study found heavy metals (lead, arsenic, cadmium) in multiple tampon brands
2023 studies detected PFAS ("forever chemicals") in menstrual products, even those marketed as PFAS-free
Vaginal and vulvar tissues absorb chemicals more rapidly than skin
Yet in the EU, manufacturers aren't required to:
Disclose ingredients on packaging
Test for specific contaminants beyond what GPSR mandates
Follow medical device biocompatibility protocols
Maintain cleanroom production environments
Conduct proactive post-market surveillance
The US requires slightly more (TSS warnings, tampon absorbency labeling) but doesn't mandate ingredient disclosure either, and the FDA only recommends (not requires) testing for pesticides, herbicides, dioxin, and specific pollutants.
The Bottom Line
Irrespective of regulatory stance, what matters is that menstrual products are produced safely and responsibly.
Kudos to Daye, Valentina Milanova, and Lisa Rodwell (ex-CEO) for pushing a whole sector towards higher standards. When a company voluntarily adopts medical device manufacturing standards for products not legally required to meet them, it demonstrates what's possible.
But we shouldn't rely on voluntary adoption. When a product is inserted into highly absorbent mucosal membranes for hours at a time, thousands of times over decades—shouldn't that merit the same regulatory rigor as a plaster on your skin?
The EU's chemical safety legislation provides strong horizontal protection. The US's medical device classification provides structural accountability. Both could learn from each other.
And both could learn from companies like Daye: when you manufacture for the body you actually have—not the regulatory minimum—everyone who menstruates benefits.
References
Linkedin Post: Daye’s cleanroom facility
Methodology Note: This article is based on my original LinkedIn post, reflecting my professional experience in femtech regulation and research into EU and US menstrual product regulatory frameworks. Claude AI assisted in elaborating the post into an article by integrating personal notes, literature research, fact-checking and further insights on the topic. All analysis and regulatory perspectives are my own, and all content has been reviewed by me for accuracy.
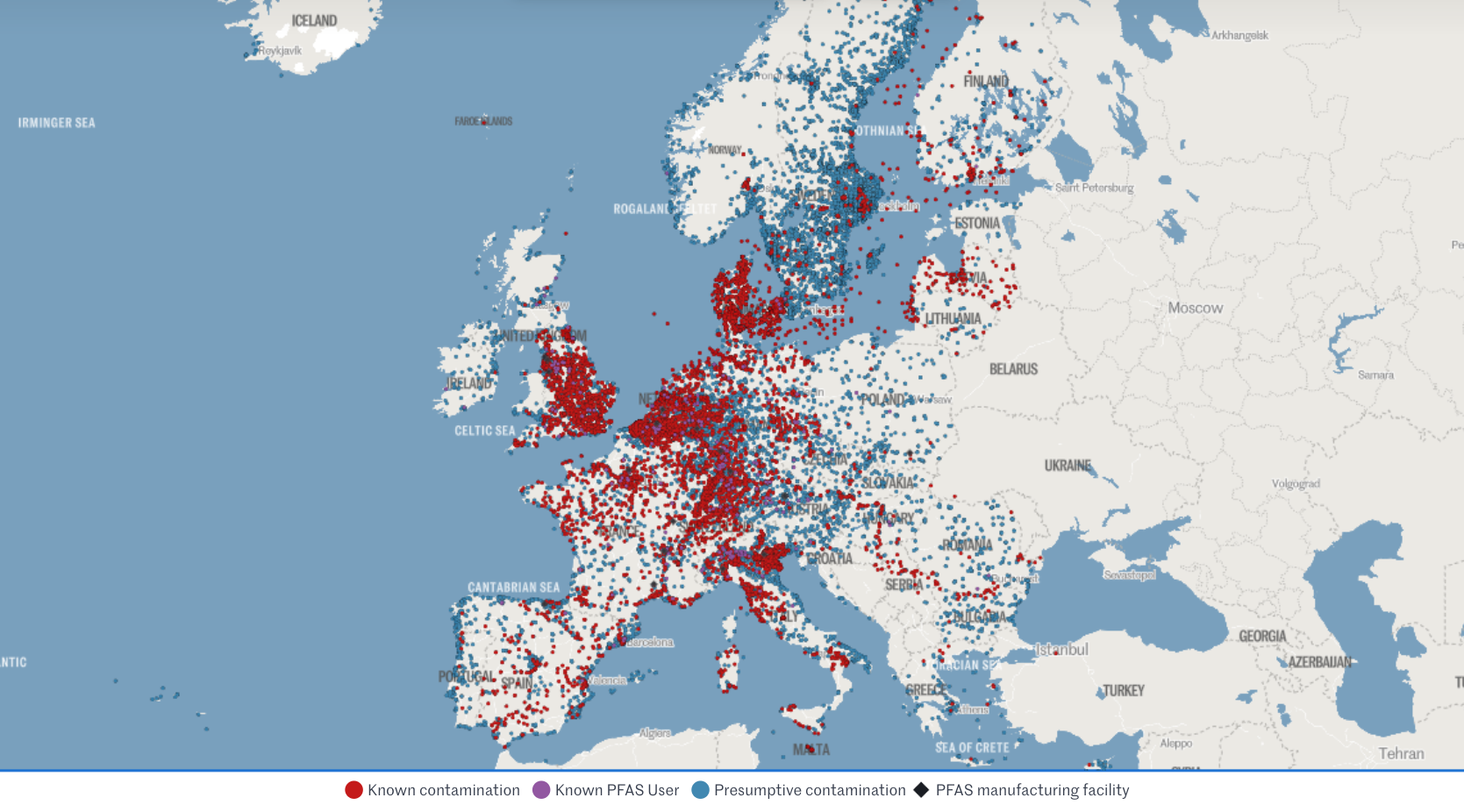
PFAS: wait-and-see Vs precautionary principle
When data is limited, do you default to safety or wait for proof of harm? This post explores the widening regulatory gap between the US and EU regarding PFAS - the "Forever Chemicals."
If you had to decide whether something is safe based on limited data, which way would you default?
Let's look at recent regulatory developments re "Teflon-like" chemicals (PFAS) in cosmetics and medical devices. Per- and polyfluoroalkyl substances (PFAS) are highly inert synthetic chemicals which makes them sought after for both everyday uses and specialist ones. However, they are so inert that biology cannot break them down. They persist in the environment and accumulate in creatures at the top of the food chain: us.
The regulatory approach to PFAS, also called Forever Chemicals, is another staggering example of the US vs. EU cultural divide.
U.S. wait-and-see approach
🇺🇸 Context: In 2024, FDA launched the Modernization of Cosmetics Regulation Act (MoCRA) which required registration of all cosmetics and listing of all their ingredients. This allowed FDA a fresh overview on PFAS' use in cosmetics, which inspired recent research.
🇺🇸 Research: A December 2025 report revealed that 51 types of PFAS are intentionally used in 1,744 cosmetic formulations in the US, commonly in makeup and even baby products.
🇺🇸 Conclusion: Due to a lack of critical toxicological data and acute toxicity, the safety of 76% of these compounds could not be definitively established. FDA deemed current evidence insufficient to justify a federal ban, opting instead for continued monitoring.
🇺🇸 Note: The FDA excluded environmental considerations and the assessment of unintentional degradation products, which are often the most harmful (e.g., PFOA and PFOS).
EU precautionary principle
🇪🇺 Context: The EU is already phasing out PFAS over concerns regarding long-term health effects and environmental contamination.
🇪🇺 Research: Rising concentrations in water streams and human blood (even in teenagers) are increasingly suspected to suppress the immune system and increase risks of cancer, infertility, thyroid dysfunction, and metabolic dysregulation.
🇪🇺 Conclusion: Action and monitoring stepped up at national and union level.
> This month, France has banned PFAS in all cosmetics (as well as clothing textiles and ski waxes).
> Yesterday, the European Environment Agency (EEA) kicked off a mandatory EU-wide program to systematically monitor PFAS in drinking water.
> Meanwhile, European Chemicals Agency (ECHA) is evaluating a proposal to ban 10,000 PFAS as a broad category, with stricter concentration limits (ppb levels) expected by October 2026.
🇪🇺 Note: The EU had already restricted all PFAS and even banned some under the REACh and the POPs regulations (which also impact allowed limits in medical devices under MDR).
Which side would you take? Personally, I’m leaning EU on this one.
Sources:
- FDA’s report
- EEA programme
- Forever pollution project (image credits)

Crans-Montana, a compliance perspective
In the wake of the devastating NYE fire in Crans-Montana, this post reflects on the critical role of compliance and individual accountability in preventing national tragedies, reminding us that regulation is only a burden until the moment it becomes our last line of defense.
Regulation is often seen as pain in the neck… until it isn't. A national tragedy takes place in Switzerland on NYE, and we ask ourselves why didn’t this underground bar have compliant emergency exits? Why wasn’t it inspected in more than 5 years? How could a combustible soundproofing material be permitted and line the whole ceiling? How could staff pull off such a deadly stunt (regularly!) with zero awareness about fire risk? Why the heck were the victims-to-be filming instead of fleeing??
And in particular, how could ALL these hazards manifest simultaneously??
I am horrified by the incident in Crans-Montana (news article). It should never have been. It lights up the painful memory of the Grenfell tower fire in 2017, which had shocked me deeply as I was living in London back then.
We all assume and expect to be protected by regulation. We all assume and expect compliant and responsible behaviour of others. The reality is that if things go south, we are on our own to face the consequences. We all have a responsibility to do our bit, whether it’s fire safety or health.
Being alert to risks, and raising the awareness of others too. Informing yourself and doing your best at least, not ignoring. Holding others accountable by asking questions or reporting unsafe practices. Raising your voice to policy-makers if something isn't enough.
I hope my work does a bit on all these things, within the realm of healthtech, of course, not fire regulation.
As a result, Switzerland now banned the use of pyrotechnics in indoor spaces and is investigating not only the bar owners but the municipality, that did not inspect the bar ONCE in 5 years. The sale of any flammable soundproofing materials is also under scrutiny.
Could this bring into 2026 a bigger wave of respect for regulation and compliance? Am I hopelessly wishful?
Today in Switzerland is a national day of mourning for the 40 victims, mostly teenagers. It breaks my heart to think of what’s left of the 116 injured.
I pray for them and for something like this to not be allowed to happen again - by regulators, by business owners, by fellow citizens, by luck (that's a factor too..🍀), by us all doing our little responsible part in society.
(Image rights: https://www.bbc.com/news/articles/c9dvyyjyj18o)

FDA’s new guidance on general wellness
This post critiques the widening regulatory gap between the US and EU following the FDA's new wellness guidance, highlighting how lower barriers for bold health claims in the US may sacrifice essential quality drivers and complicate global strategies for startups.
Yesterday's release by FDA on wellness vs medical device leaves me with a bitter aftertaste. Why?
I'm usually enthusiastic about policies that lower the barrier to market entry for health products. I'm less enthusiastic about those that eliminate the quality drivers from it..
My main concerns under this guidance:
> General wellness products have no QMS requirement, especially digital ones. So when the guidance says you can now display biomarkers even with some disease reference as long as "the product has validated values" for those biomarkers, it doesn't really mean anything. How do they validate? According to what? Where? Claims get bolder and accountability weaker.
> We will see more products being Class IIa medical devices in EU (with QMS auditing and device file review) while facing zero expectations in the US as general wellness.
> The gap between EU and US regulatory approach gets wider. EU released a "similar" guidance in Sep 2025 emphasising the opposite, with increased focus on mechanism of action and technology rather than relying on claims only. US heads the other way, making it all the more complicated for us RA 🥴
> It will be harder for startups to design their product and strategies for the two main western markets simultaneously. They will be pushed even heavier towards wellness-first but in my experience they get easily stuck there.
> This bold approach may be (too) specific of this administration. Will it then outlive it? It is also clearly result from the WHOOP controversy, given the number of references to Blood Pressure measuring wrist-worn devices. Pretty solid legal and lobby teams there.
One example that puzzles me in particular is the one about glucose monitoring via "minimally invasive microneedle technology" for which FDA says they will apply enforcement discretion as a low risk device. Since I'm currently working on the biocompatibility testing requirements for a device that is hand held by doctors using gloves (👀), I cannot help but finding it unfair towards the rest of the sector.
So I hope you will excuse my slightly less upbeat post this time.
I'm generally excited about the expansion of the definition and agree with the rationale of most of the examples provided.
I'm curious to see what it will mean for international harmonisation and for the opportunities it will open for my clients at this interface!

MDR/IVDR proposal for simplication
This post highlights the European Commission's groundbreaking proposal to overhaul and simplify the MDR and IVDR frameworks, promising more proportionate rules for low-risk devices, reduced administrative burdens for SMEs, and a modern, digital-first approach to medtech regulation in the EU.
12 hours ago the European Commission published THE MOST AWAITED AND CRUCIAL DEVELOPMENT IN A DECADE: its proposal for simplification of the MDR and IVDR. 👏
Alert: it is still only a proposal, albeit official, which has been submitted to the European Parliament and the Council, but will need to go through the ordinary legislative procedure to become binding Union law.
From a first diagonal read, what struck my attention:
🎉 More room for Class I devices, incl software (THANK YOU!)
🎉 Simplified interaction with AI Act
🎉 Codified instruments for open dialogue on classification and access to expert panels
🎉 Easier "equivalence" concept including use of synthetic data,
🎉 Lower NB fee structure for SMEs
🎉 Extended reporting timelines and validity of certificates
🎉 Reduced scope of surveillance audits and conformity assessment
🎉 Built-in flexibility for public health emergencies, breakthrough/orphan devices (i.e. life-threatening, rare, untreated diseases), supply-chain disruptions
Interestingly, but unsurprisingly, it proposes additional requirements for cybersecurity conformity and reporting (beyond what qualifies as medically "serious").
I will share more details of how this would impact specifically medical device startups especially in digital health and femtech.
While it is still ONLY A PROPOSAL, it is sign that EU is listening and actively working to "make [the current rules] easier, faster and more effective and further promote competitiveness, innovation and a high-level of patient safety in this key sector"
We're excited to follow the development of the legislative decision-making process and wait eagerly for the change of an era this (or its variants that will result) will bring to the European medtech sector!

What can we learn from… Canada?
This post explores the "Canadian Technology Accelerator" model for international expansion, sharing insights from a bilingual FemTech panel in Paris on how high-potential startups - like PCOS-focused mentee Élan Healthcare - can leverage diplomatic networks and local mentorship to navigate global regulatory compliance.
What am I doing sitting in a bilingual English-French panel at the Embassy of Canada | Ambassade du Canada in Paris??
Talking femtech regulatory compliance trends (in English) while listening (mostly in live-translated French) to the perspectives of brilliant entrepreneurs, investors, researchers and diplomats!
This would be for my series of "What can we learn from... Canada?"
The Canadian Trade Commission runs the Canadian Technology Accelerators | Accélerateurs technologiques canadiens, a programme to support Canadian startups to expand to other markets. By collaborating with global Canadian embassies they provide eligible startups with local mentorship, contacts and partnerships to boost their growth.
As part of this, I had the privilege to mentor Élan Healthcare Inc. run by Pari (Parvaneh) Saharkhiz, MD, MBA, a doctor turned founder and manufacturer of supplements especially designed to tackle the nutritional imbalances that are often root to PCOS and infertility. Around 10% of women are affected by PCOS, 70% go undiagnosed, and even those who have it diagnosed struggle to find treatment. Check them out: https://elanhealthcare.ca/
Grateful for the invitation to Trade Commissioner Frederic Chieux and Fiona Thwaites. A pleasure to sit on the panel with collaborator, friend and amazing host Erica Perrier, PhD, MS, CSCS as well as great copanelists Régine Brielle Juliette Mauro Andrea Guest Andréa Saragoussi Keshiv Kaushal - thanks for sharing your knowledge.
Greatest success to the impressively advanced startups in the mentee cohort Cogni Cosm Medical Emovi Juno Technologies™ Mino Care My Normative LoOoP SYNG Pharmaceuticals Inc, I look forward to staying in touch!

What can we learn from… a progressive Notified Body?
Medtech governance in Europe is highly decentralised, with product certifications also being "outsourced" to private entities (i.e. Notified Bodies). This would be complicated enough if classic Notified Bodies didn't also bring their own enormous challenges to the table: lack of availability, lack of new tech competence, lack of transparency and communication.. Companies feel they have no control over their destiny.
So what's Scarlet doing differently as a Notified Body:
1️⃣ Focus on one subject matter (digital devices only) to ensure top and uptodate competence
2️⃣ Fit the conformity assessment process around the applicant and their timelines
3️⃣ Engage transparently and pragmatically about expectations in pre-sub Structured Dialogues
4️⃣ Scale resources flexibly with externals
and, my favourite,
5️⃣ Train their trusted consultants in an independent manner in order to increase the chance of high quality submissions and enable more effective reviews.
Which other NBs do this? None that I'm aware. But please share if you know any good practices you've experienced.
Therefore, I'm particularly enthusiastic to have been part of this special training session last Friday! Not only with a like-minded NB, but among a group of 18 like-minded regulatory experts ❤️
New times and new tech need a new approach - a mantra of Edge Compliance. I hope other and new NBs will take example.
Note: I'm not affiliated but believe the initiative deserves genuine praise and broadcasting.
Thank you Dan Levy and Sandy Wright at Scarlet - also for the photo credit. Stellar job!

PCCP beyond AI
Very exciting trend of femtech apps integrating with wearable data! How does this work for the regulated ones? I wanted to share this clever use of PCCP from Natural Cycles° from last year which impressed me.
What's PCCP?
Pre-determined Change Control Plan is a regulatory instrument devised by FDA - as a European is I'm most jealous of. It was designed to enable AI devices, which by design need to be able to evolve their accuracy in the field, getting smarter the more data they acquire. Traditionally, any change to the accuracy and performance of a device required a regulatory resubmission (still the case in EU) and up to 90 days of review wait.
With PCCP you can get pre-approval for a reasonable range of performance that you anticipate and accept.
What I found clever, is that Natural Cycles°, the pioneer of regulated fertility awareness, used PCCP not for AI changes but for variability of source data from different wearables.
While, as far as I'm aware, they currently integrate only with ŌURA and Apple Watch, this clears the way for them to swiftly add any more integrations to their conception/contraception suite as long as they fit their predefined specs (see table in pdf).
This is an example of how:
1️⃣ Regulatory instruments that are smart and abreast with the times enable even more innovation than what they primarily intended to,
2️⃣ Femtech is riding the wave of biomarkers ensuring most users can be served irrespective of which devices they choose - it's not just the iOS vs Android divide anymore!
3️⃣ Scientific research and clinical partnerships will see an incredible boost of opportunity from all this data, finally compensating for the lack of data that we know womens health has suffered until now!
What else could we use PCCP for? And until when can we have a similar toolkit in Europe under MDR? 🫠
NC's current integrations here
Link to full 510k summary here

What can we learn from… Australia?
What if the interaction with regulators was more personal?
This week, I was interviewed as part of the Therapeutic Goods Administration's research for improving health software regulation.
Since we registered a SaMD client in Australia, we were contacted to take part in a 1 hour call with ORIMA Research on the TGA's behalf. We had the chance to discuss our experience and to give suggestions on what would help other digital health companies enter the Australian market compliantly, for example:
🔍 how do companies find out whether they are regulated?
🔍 what is key for them to know in order to navigate the regs?
🔍 what's clear / unclear in the regs?
🔍 what could be attracting digital health companies to Australia?
🔍 what would put them off from doing it compliantly?
I would love to see the EU doing the same. But then, in practice, who? The European Commission? The national Competent Authorities? The Notified Bodies? Team-NB? MDCG? It gets complicated before even starting..
Yes, sometimes the EC issues calls for comments on certain regulations. The problem I have with this is a) the free-text format, which is an invite for whining and venting, and b) the lack of accountability, i.e. does anyone read it? what happens with it?
In contrast, I really appreciated:
🌟 The structured discussion format, still with some liberty to digress,
🌟 The face-to-face personal interaction, which encourages trust,
🌟 Knowing a report with clear actions will come out of it and disseminated.
Kudos to the TGA and lovely experience chatting to Jack Disher at ORIMA.
We look forward to the report!

Regulation without borders
Starting two new client projects this week, one on food supplements in France and one on in-vitro diagnostics in Germany, both in womens health!
Very few medtech consultants would feel comfortable touching other verticals (even from MDR to IVDR). But my career started like that when, honestly, I didn't have a choice! Now it's what I enjoy the most, and what I built my agency around.
The hard competences boil down to a few common traits, irrespective of sectors, regs and countries:
➡️ Regulatory definition / classification
➡️ Manufacturing requirements
➡️ Claims and label compliance
➡️ Responsible Person / Entity role
➡️ Notification / Submission procedures
➡️ Review interaction
➡️ Launch and Distribution
➡️ Post-market reporting
After all, it's all about health accountability, and humans have really one way of expecting it - the rest is often noise.
Personally, I find it super fun to come across these analogies, transfer learnings from one area to another and even anticipate cross-sector currents. Excited to get going!

At Women’s Health Week Europe
The go-to regulatory compliance agency for femtech. Find out more.
An absolutely incredible time at Women's Health Week at the Barbican! The opportunity and momentum for femtech re undeniable.
It's no philanthropic initiative, it's a business sector with real problems be solved and real money to be made. Women, i.e. 51% of the population, control more than 80% of household health decisions, yet suffer ill-health for 19% more time than men - mostly during their working years.
Closing the gap could generate >$1trillion annual global GDP by 2040. It's no charity, it's business sense.
The innovation that is happening in this space fills us with awe, anticipation and gratitude. Will post more about some of the founders and products that we learnt about.

Review timelines for FDA 510k clearance
How long does it take from FDA submission to clearance?
Let's look at the recent data.
The 510k database can be exported and analysed. Format is not humanly readable but makes a fun ChatGPT exercise.
Here is the result of me playing with the database from devices cleared last months (Aug and Sep 2025).
❗ The normal distribution appears to peak around 90 days, the legal obligation for FDA to respond to submissions. Around 30% of submissions were cleared within that timeframe.
❗ Nice peak at 30 days - but don't be too wishful! These are expedited reviews, e.g. changes to existing 510ks or based on prior agreements or expected updates.
❗ Less exciting peak around 270 days, i.e. 9 months. Most submissions receive an Additional Information request, which gives manufacturers 180 days to respond and restarts the clock for FDA after that (further 90 days).
Lesson here?
If you're planning a 510k, a realistic estimate for clearance is nothing less than 6 months. This is what applied to 2/3s of the 400+ applications cleared most recently.
Good quality submissions and preliminary discussions with FDA on the fundamental topics can help prevent Additional Information requests and thus increase the chances of receiving clearance within 90 days.
Does your experience confirm this too?
I will dig more into this database in the coming posts with more insights.

US Gov shutdown: impact on FDA operations
After Republican and Democratic politicians could not agree to pass a bill funding government services, on 1st October the US federal government has shut down. Though not unusual (almost every administration had at least one, lasting from a couple of days to a top 35 days), they create immediate uncertainty for largely Congress-funded agencies such as the FDA.
FDA announced that, based on its contingency plan, it will limit its ops to “mission critical activities including responding to public health emergencies, supporting high-risk food and medical product recalls, and conducting essential surveillance of medical devices and other medical products”.
So in practice, until the end of the shutdown:
🔴 No new submissions accepted (510k, DeNovo) nor payments thereof,
🟡 Ongoing reviews will continue but may suffer delays beyond the mandatory timeframes and potential unresponsiveness,
🔴 Annual fees will not be processed (MDUFA user registration), though due in October for Fiscal Year 2026 - see my previous post on increased fees,
🟢 Medical device recalls and safety surveillance will continue,
🟡 Inspections largely on hold except if “for cause”.
Tough news if you are on the brink of submitting or awaiting a decision. But history tells us these don't last long, so be ready to move fast once the shutdown lifts.

Is it cake? New bordeline guideline rundown
Here the regulatory version of “IS IT CAKE??” 🍰 - if you know the show! Featuring the European Commission’s updated guidance on borderline products published this month.
As someone whose specialty is borderline products and who loves RA developments on the edge, I spent hours digesting its 24 examples of what is or isn’t a medical device - as opposed to drugs, cosmetics, IVD, personal protective equipment (PPE), biocides,..
Frankly, I found half of the examples straightforward, and the other half.. I either struggle to understand the reasoning, disagree or find it inconsistent. Here the main reasons:
INTENDED USE vs MODE OF ACTION, WHO WINS?
MDR defines and classifies medical devices based on the former, while this guidance mostly hedges on the latter. When conflicts arise, this guidance gives priority to the mode of action. There are two, in my opinion, conflicting examples with devices that claim prevention of disease: an STI prevention app and medical examination table covers (i.e. paper roll). The first is not MD, despite processing medical records, using algorithms to assess risk, alerting peers regarding their potential for infection - because “no action on data other than communication”. The second is MD, regardless of its make - because “acts as a mechanical barrier”.
ANYTHING BUT Class I, EVER..
The myth of Class I devices continues. Only one example from here comes out as Class I MD: a rescue bag for patient transport - because "aims to support and protect, [..] avoids worsening of health". Arguably, PPE and Product for Emergency Rescue regulations could be sufficient, so what does Class I MD status really add here? On the other hand, why couldn’t some other low risk examples be Class I (e.g. STI app above, medical calculator for recurrent math)?
It's a continuous learning process for all, and access to practical guidance of this type is very helpful for the health sector as a whole - actually something that FDA does way better (writing style and formal consistency in this manual is quite disappointing).
If we held a geeky RA pub quiz on these examples, how would RA professionals, national authorities and notified bodies score? That would be interesting.

At Swiss Medtech 2025
Swiss Medtech events never disappoint!
Key learnings from attending yesterdays session in sunny Bern (inside a stunning casino!):
1️⃣ US tariffs and lower FDA capacity are discouraging EU/CH startups from going US-first, but there are clever best-practices to work around them.
2️⃣ EU's gap between numbers in MDR applications and certifications is widening in unsustainable ways due to a poor EU-wide governance model for medtech, and how this needs fixing ASAP.
3️⃣ Switzerland is working out creative legal basis to be an attractive alternative (e.g. to fast-track FDA medical devices and to modernise its regulatory framework faster than the EU can)
4️⃣ Emerging markets (e.g. Saudi Arabia) get devices to market 6 months faster than traditional markets, meaning their patients get better outcomes, HCPs get better education, and the healthcare system innovates exponentially faster.
Grateful to Bernhard Bichsel and Sandra Item from ISS AG, Integrated Scientific Services, Daniel Delfosse, Eva von Mühlenen, LL.M., from Sidley Austin LLP, Glenda C. Marsh from Johnson & Johnson MedTech for putting together such an inspiring and informative afternoon!
LLM for Quality tasks
A short story on using AI for a QARA task and coming up with a framework for doing it faster (4h down to 1h) while keeping it under control.
Task at hand:
Client received its inspection report from the authority via the post in the national language and needed it digitalised and in English in order to action it.
1️⃣ Convert scanned pdf to electronic document
ChatGPT 👎 didn’t identify text in the scanned pdf.
Gemini and NotebookLM did it, but I was unconvinced by the accuracy 🧐 .
GoogleDrive did the job, uploaded the pdf and "Open as GoogleDoc". ✅
2️⃣ Translate electronic document
ChatGPT and Gemini kept hallucinating badly 😵💫 .
The "Translate document" function of GoogleDocs returned a poor literal translation 🥴 .
NotebookLM was accurate but skipped content 😥 .
Ended up doing section by section via Gemini's in-text "AI Refine" function with a very meticulous prompt and checking it manually in a side-by-side table 🥵 .
3️⃣ Format electronic document similar to the original
ChatGPT and NotebookLM didn’t work 🤕 .
Gemini could do some basic improvements via the in-text "AI Refine" function, but not via the GoogleDocs built-in "Ask Gemini" nor via the browser chat. Interesting how much these differ in capability.
In the end, the formatting fix was mostly manual 🤯 .
Conclusion:
After 4 miserable hours spent on the task with many failed attempts and much too manual input, I achieved a satisfactory document.
But, I still wanted to get to the bottom of this. There must be a better way??
So I restarted from scratch using a different approach, which I could summarise in a way that is inspired by the concept of the PDCA / Agile cycle we use in Quality:
⤵️ Plan: Ask AI for the right tools and prompts to achieve your goal. And importantly, "ask AI to ask you" questions or point out what is unclear in order to help you refine your requirements accurately.
▶️ Do: Approach it step by step. Run your refined prompt for your SUBtask in your selected tool. Quick review of the output, refine the prompt. Change tool if needed.
⏯️ Check: Get AI to verify its results and to help you check it manually by highlighting any discrepancies. For example, “juxtapose the original and translated content in a table section by section and note any discrepancies between the two version of the text”.
🔁 Act: Tell AI to correct the discrepancies, then re-run the verification step to update results.
Eventually, by doing it this way, I could achieve the same result in 1h and with increased confidence on the accuracy. Still not extremely fast, but considerably faster!
I am curious, how would others have approached this dull task?