
PFAS: wait-and-see Vs precautionary principle
When data is limited, do you default to safety or wait for proof of harm? This post explores the widening regulatory gap between the US and EU regarding PFAS - the "Forever Chemicals."
If you had to decide whether something is safe based on limited data, which way would you default?
Let's look at recent regulatory developments re "Teflon-like" chemicals (PFAS) in cosmetics and medical devices. Per- and polyfluoroalkyl substances (PFAS) are highly inert synthetic chemicals which makes them sought after for both everyday uses and specialist ones. However, they are so inert that biology cannot break them down. They persist in the environment and accumulate in creatures at the top of the food chain: us.
The regulatory approach to PFAS, also called Forever Chemicals, is another staggering example of the US vs. EU cultural divide.
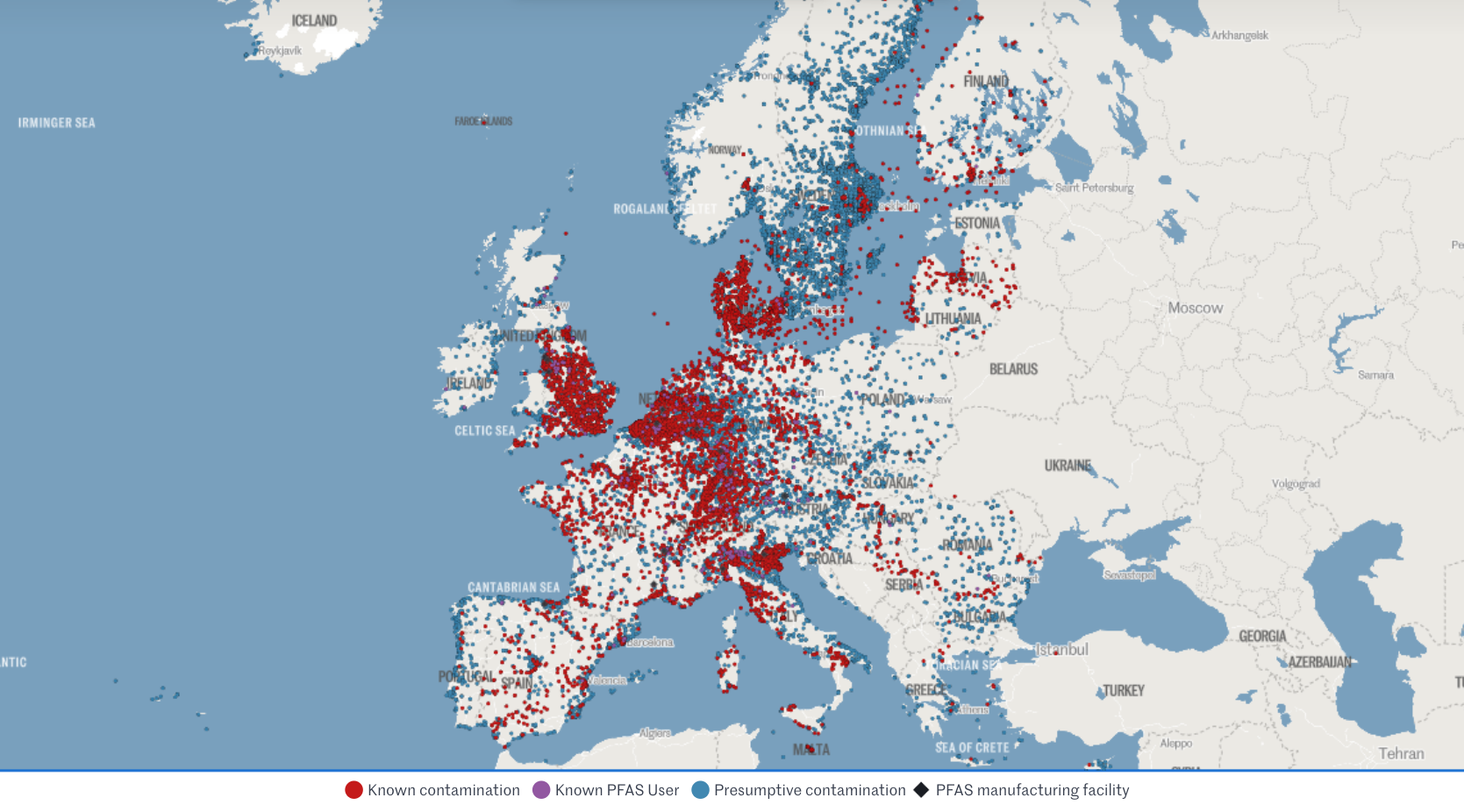
U.S. wait-and-see approach
🇺🇸 Context: In 2024, FDA launched the Modernization of Cosmetics Regulation Act (MoCRA) which required registration of all cosmetics and listing of all their ingredients. This allowed FDA a fresh overview on PFAS' use in cosmetics, which inspired recent research.
🇺🇸 Research: A December 2025 report revealed that 51 types of PFAS are intentionally used in 1,744 cosmetic formulations in the US, commonly in makeup and even baby products.
🇺🇸 Conclusion: Due to a lack of critical toxicological data and acute toxicity, the safety of 76% of these compounds could not be definitively established. FDA deemed current evidence insufficient to justify a federal ban, opting instead for continued monitoring.
🇺🇸 Note: The FDA excluded environmental considerations and the assessment of unintentional degradation products, which are often the most harmful (e.g., PFOA and PFOS).
EU precautionary principle
🇪🇺 Context: The EU is already phasing out PFAS over concerns regarding long-term health effects and environmental contamination.
🇪🇺 Research: Rising concentrations in water streams and human blood (even in teenagers) are increasingly suspected to suppress the immune system and increase risks of cancer, infertility, thyroid dysfunction, and metabolic dysregulation.
🇪🇺 Conclusion: Action and monitoring stepped up at national and union level.
> This month, France has banned PFAS in all cosmetics (as well as clothing textiles and ski waxes).
> Yesterday, the European Environment Agency (EEA) kicked off a mandatory EU-wide program to systematically monitor PFAS in drinking water.
> Meanwhile, European Chemicals Agency (ECHA) is evaluating a proposal to ban 10,000 PFAS as a broad category, with stricter concentration limits (ppb levels) expected by October 2026.
🇪🇺 Note: The EU had already restricted all PFAS and even banned some under the REACh and the POPs regulations (which also impact allowed limits in medical devices under MDR).
Which side would you take? Personally, I’m leaning EU on this one.
Sources:
- FDA’s report
- EEA programme
- Forever pollution project (image credits)

FDA’s new guidance on general wellness
This post critiques the widening regulatory gap between the US and EU following the FDA's new wellness guidance, highlighting how lower barriers for bold health claims in the US may sacrifice essential quality drivers and complicate global strategies for startups.
Yesterday's release by FDA on wellness vs medical device leaves me with a bitter aftertaste. Why?
I'm usually enthusiastic about policies that lower the barrier to market entry for health products. I'm less enthusiastic about those that eliminate the quality drivers from it..
My main concerns under this guidance:
> General wellness products have no QMS requirement, especially digital ones. So when the guidance says you can now display biomarkers even with some disease reference as long as "the product has validated values" for those biomarkers, it doesn't really mean anything. How do they validate? According to what? Where? Claims get bolder and accountability weaker.
> We will see more products being Class IIa medical devices in EU (with QMS auditing and device file review) while facing zero expectations in the US as general wellness.
> The gap between EU and US regulatory approach gets wider. EU released a "similar" guidance in Sep 2025 emphasising the opposite, with increased focus on mechanism of action and technology rather than relying on claims only. US heads the other way, making it all the more complicated for us RA 🥴
> It will be harder for startups to design their product and strategies for the two main western markets simultaneously. They will be pushed even heavier towards wellness-first but in my experience they get easily stuck there.
> This bold approach may be (too) specific of this administration. Will it then outlive it? It is also clearly result from the WHOOP controversy, given the number of references to Blood Pressure measuring wrist-worn devices. Pretty solid legal and lobby teams there.
One example that puzzles me in particular is the one about glucose monitoring via "minimally invasive microneedle technology" for which FDA says they will apply enforcement discretion as a low risk device. Since I'm currently working on the biocompatibility testing requirements for a device that is hand held by doctors using gloves (👀), I cannot help but finding it unfair towards the rest of the sector.
So I hope you will excuse my slightly less upbeat post this time.
I'm generally excited about the expansion of the definition and agree with the rationale of most of the examples provided.
I'm curious to see what it will mean for international harmonisation and for the opportunities it will open for my clients at this interface!

MDR/IVDR proposal for simplication
This post highlights the European Commission's groundbreaking proposal to overhaul and simplify the MDR and IVDR frameworks, promising more proportionate rules for low-risk devices, reduced administrative burdens for SMEs, and a modern, digital-first approach to medtech regulation in the EU.
12 hours ago the European Commission published THE MOST AWAITED AND CRUCIAL DEVELOPMENT IN A DECADE: its proposal for simplification of the MDR and IVDR. 👏
Alert: it is still only a proposal, albeit official, which has been submitted to the European Parliament and the Council, but will need to go through the ordinary legislative procedure to become binding Union law.
From a first diagonal read, what struck my attention:
🎉 More room for Class I devices, incl software (THANK YOU!)
🎉 Simplified interaction with AI Act
🎉 Codified instruments for open dialogue on classification and access to expert panels
🎉 Easier "equivalence" concept including use of synthetic data,
🎉 Lower NB fee structure for SMEs
🎉 Extended reporting timelines and validity of certificates
🎉 Reduced scope of surveillance audits and conformity assessment
🎉 Built-in flexibility for public health emergencies, breakthrough/orphan devices (i.e. life-threatening, rare, untreated diseases), supply-chain disruptions
Interestingly, but unsurprisingly, it proposes additional requirements for cybersecurity conformity and reporting (beyond what qualifies as medically "serious").
I will share more details of how this would impact specifically medical device startups especially in digital health and femtech.
While it is still ONLY A PROPOSAL, it is sign that EU is listening and actively working to "make [the current rules] easier, faster and more effective and further promote competitiveness, innovation and a high-level of patient safety in this key sector"
We're excited to follow the development of the legislative decision-making process and wait eagerly for the change of an era this (or its variants that will result) will bring to the European medtech sector!

What can we learn from… Canada?
This post explores the "Canadian Technology Accelerator" model for international expansion, sharing insights from a bilingual FemTech panel in Paris on how high-potential startups - like PCOS-focused mentee Élan Healthcare - can leverage diplomatic networks and local mentorship to navigate global regulatory compliance.
What am I doing sitting in a bilingual English-French panel at the Embassy of Canada | Ambassade du Canada in Paris??
Talking femtech regulatory compliance trends (in English) while listening (mostly in live-translated French) to the perspectives of brilliant entrepreneurs, investors, researchers and diplomats!
This would be for my series of "What can we learn from... Canada?"
The Canadian Trade Commission runs the Canadian Technology Accelerators | Accélerateurs technologiques canadiens, a programme to support Canadian startups to expand to other markets. By collaborating with global Canadian embassies they provide eligible startups with local mentorship, contacts and partnerships to boost their growth.
As part of this, I had the privilege to mentor Élan Healthcare Inc. run by Pari (Parvaneh) Saharkhiz, MD, MBA, a doctor turned founder and manufacturer of supplements especially designed to tackle the nutritional imbalances that are often root to PCOS and infertility. Around 10% of women are affected by PCOS, 70% go undiagnosed, and even those who have it diagnosed struggle to find treatment. Check them out: https://elanhealthcare.ca/
Grateful for the invitation to Trade Commissioner Frederic Chieux and Fiona Thwaites. A pleasure to sit on the panel with collaborator, friend and amazing host Erica Perrier, PhD, MS, CSCS as well as great copanelists Régine Brielle Juliette Mauro Andrea Guest Andréa Saragoussi Keshiv Kaushal - thanks for sharing your knowledge.
Greatest success to the impressively advanced startups in the mentee cohort Cogni Cosm Medical Emovi Juno Technologies™ Mino Care My Normative LoOoP SYNG Pharmaceuticals Inc, I look forward to staying in touch!

What can we learn from… a progressive Notified Body?
Medtech governance in Europe is highly decentralised, with product certifications also being "outsourced" to private entities (i.e. Notified Bodies). This would be complicated enough if classic Notified Bodies didn't also bring their own enormous challenges to the table: lack of availability, lack of new tech competence, lack of transparency and communication.. Companies feel they have no control over their destiny.
So what's Scarlet doing differently as a Notified Body:
1️⃣ Focus on one subject matter (digital devices only) to ensure top and uptodate competence
2️⃣ Fit the conformity assessment process around the applicant and their timelines
3️⃣ Engage transparently and pragmatically about expectations in pre-sub Structured Dialogues
4️⃣ Scale resources flexibly with externals
and, my favourite,
5️⃣ Train their trusted consultants in an independent manner in order to increase the chance of high quality submissions and enable more effective reviews.
Which other NBs do this? None that I'm aware. But please share if you know any good practices you've experienced.
Therefore, I'm particularly enthusiastic to have been part of this special training session last Friday! Not only with a like-minded NB, but among a group of 18 like-minded regulatory experts ❤️
New times and new tech need a new approach - a mantra of Edge Compliance. I hope other and new NBs will take example.
Note: I'm not affiliated but believe the initiative deserves genuine praise and broadcasting.
Thank you Dan Levy and Sandy Wright at Scarlet - also for the photo credit. Stellar job!

Regulation without borders
Starting two new client projects this week, one on food supplements in France and one on in-vitro diagnostics in Germany, both in womens health!
Very few medtech consultants would feel comfortable touching other verticals (even from MDR to IVDR). But my career started like that when, honestly, I didn't have a choice! Now it's what I enjoy the most, and what I built my agency around.
The hard competences boil down to a few common traits, irrespective of sectors, regs and countries:
➡️ Regulatory definition / classification
➡️ Manufacturing requirements
➡️ Claims and label compliance
➡️ Responsible Person / Entity role
➡️ Notification / Submission procedures
➡️ Review interaction
➡️ Launch and Distribution
➡️ Post-market reporting
After all, it's all about health accountability, and humans have really one way of expecting it - the rest is often noise.
Personally, I find it super fun to come across these analogies, transfer learnings from one area to another and even anticipate cross-sector currents. Excited to get going!

Is it cake? New bordeline guideline rundown
Here the regulatory version of “IS IT CAKE??” 🍰 - if you know the show! Featuring the European Commission’s updated guidance on borderline products published this month.
As someone whose specialty is borderline products and who loves RA developments on the edge, I spent hours digesting its 24 examples of what is or isn’t a medical device - as opposed to drugs, cosmetics, IVD, personal protective equipment (PPE), biocides,..
Frankly, I found half of the examples straightforward, and the other half.. I either struggle to understand the reasoning, disagree or find it inconsistent. Here the main reasons:
INTENDED USE vs MODE OF ACTION, WHO WINS?
MDR defines and classifies medical devices based on the former, while this guidance mostly hedges on the latter. When conflicts arise, this guidance gives priority to the mode of action. There are two, in my opinion, conflicting examples with devices that claim prevention of disease: an STI prevention app and medical examination table covers (i.e. paper roll). The first is not MD, despite processing medical records, using algorithms to assess risk, alerting peers regarding their potential for infection - because “no action on data other than communication”. The second is MD, regardless of its make - because “acts as a mechanical barrier”.
ANYTHING BUT Class I, EVER..
The myth of Class I devices continues. Only one example from here comes out as Class I MD: a rescue bag for patient transport - because "aims to support and protect, [..] avoids worsening of health". Arguably, PPE and Product for Emergency Rescue regulations could be sufficient, so what does Class I MD status really add here? On the other hand, why couldn’t some other low risk examples be Class I (e.g. STI app above, medical calculator for recurrent math)?
It's a continuous learning process for all, and access to practical guidance of this type is very helpful for the health sector as a whole - actually something that FDA does way better (writing style and formal consistency in this manual is quite disappointing).
If we held a geeky RA pub quiz on these examples, how would RA professionals, national authorities and notified bodies score? That would be interesting.

EU AI Act deployment
Since August 2nd the EU AI Act is in force. But is it?
In practice: not much today, but the clock has started. If your device includes an AI component or uses AI to support decisions it’s time to take a closer look.
For high-risk systems, including many AI-based medical devices, there’s a 36-month transition to comply, i.e. phased implementation. However, some provisions apply earlier (e.g. banned uses of AI, codes of conduct).
Here’s what I see across medtech:
1. Confusion around scope and classification, e.g. AI as a tool for CSV or as part of the intended use?
2. Assumptions that MDR = AI Act compliance, thus reactive attitude to QMS updates upon NB feedback rather than in a proactive manner
3. Teams don't know how to resource it.
Good thing is that I also see a booming AI-related offering from QARA consultants and training providers which can help if you’re stuck on any of the above points. Cool examples (among many others):
• AI-first QARA frameworks and training e.g. Johner Institut GmbH https://lnkd.in/dBSuFfie,
• AI agents for compliance-checking and even FDA review outcome prediction such as Lexim AI or Acorn Compliance,
• GenAI embedded in eQMS tools such as Formwork from OpenRegulatory or Matrix One
What would help your team implementing the AI Act? Curious to hear your challenges and to help you with the right support.