
The next strategic frontiers of medtech
Where should medtech and femtech companies focus their international expansion efforts after establishing EU and US market presence? This article analyses eight high-potential markets across four global regions — Anglosphere, East Asia, MENA, and Latin America — ranked against three strategic criteria: ROI and medtech infrastructure, femtech readiness, and regulatory alignment with international harmonisation frameworks including MDSAP and CE Mark mutual recognition agreements. Whether you are mapping your next regulatory strategy or building a market entry roadmap, this evidence-based analysis offers a practical starting point — with an honest look at what the data says and where gut instinct still has a role to play.
After my popular recent poll, which strategic markets deserve to be your and my next medtech focus beyond US-EU? Note: beyond, not instead of!
What are the factors we want to optimise?
ROI & MEDTECH INFRASTRUCTURE: markets that offer the highest investment and monetization potential, with advanced payer structures and tech scaling capabilities.
FEMTECH READINESS: regions where the cultural and policy discourse is opening to women’s health, where clinical research is advanced, and femtech innovation and startup ecosystem are thriving.
REGULATORY ALIGNMENT: countries that align with international medtech harmonisation efforts, for example being members of MDSAP or with mutual recognition agreements (MRA) towards CE mark and FDA.
With the help of Claude, I ran an advanced analysis to find the right countries for these categories. More details on the results summarised below.
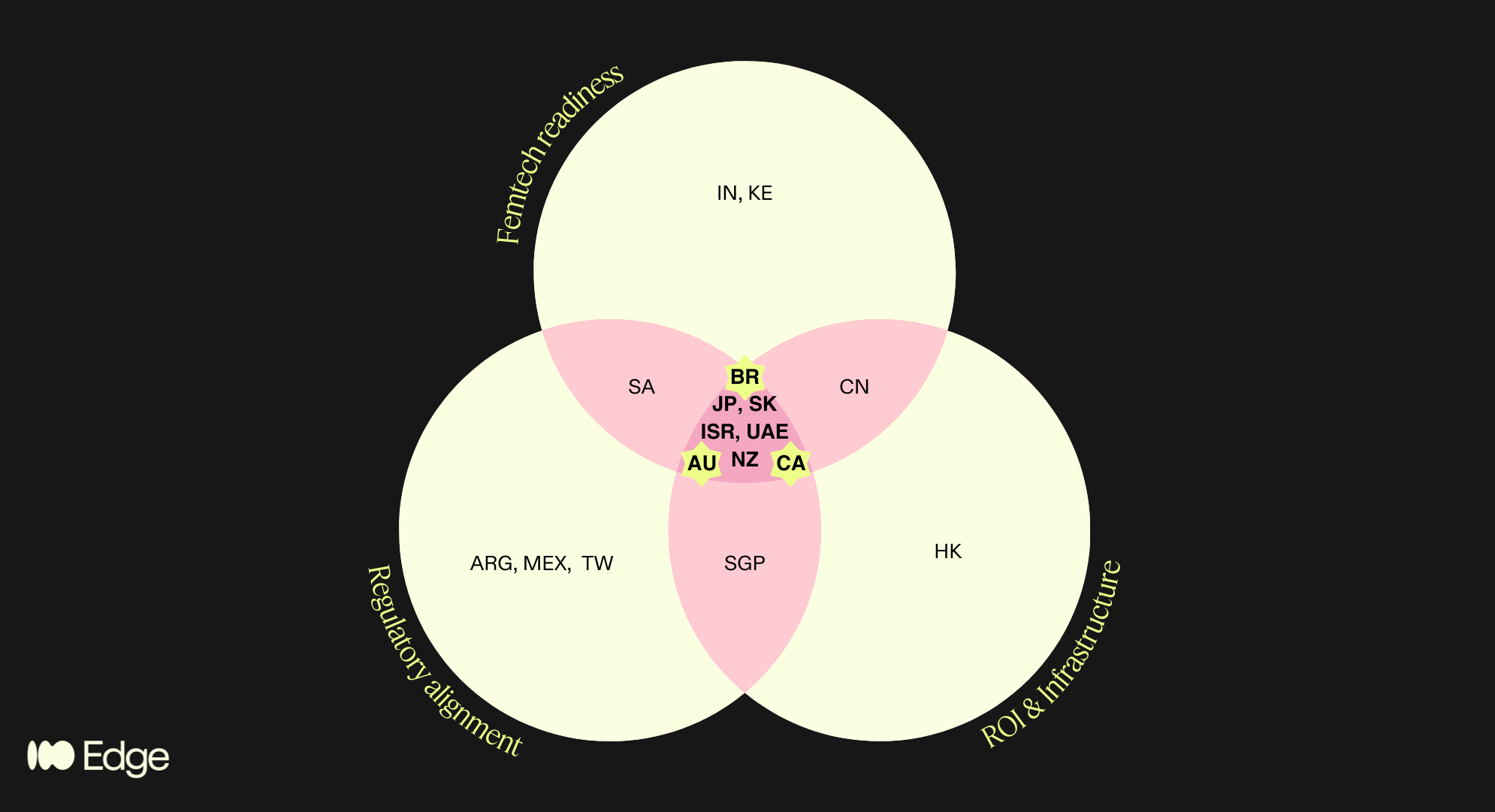
Regions that satisfied multiple criteria are highlighted in the pink areas of the diagram. This brings us to 8 high potential regions, spanning the 4 geographies of my poll!
Anglosphere: Canada, Australia, New Zealand
“BRIC”+East Asia: Japan, South Korea
MENA: Israel, UAE
LatAm: Brazil
What does it mean for your market strategy? If you are in femtech and looking to expand after EU/US proof-of-concept - AND not planning to exit directly - then these are your markets. Save and share the post, and narrow down the list based on further criteria relevant to you and your business.
To me and my business, I applied the following additional criteria:
Accessibility: where can my cultural roots and the languages I speak help me?
Market size: >20M for highest chances of leads and indirect product reach for my clients
Heterogeneity: the more diverse the better for clinical research relevance
Geopolitical stability: low risk of conflicts and market volatility
Which takes us to the winners!
🇧🇷 Brasil: as half-Brazilian I’ll be proud and excited to explore the femtech, clinical and regulatory space and build connections there! DM me to discuss!
🇦🇺 🇨🇦 Australia and Canada: it’s a tie! I already work with these markets, no big lift, but good pointer to go a bit deeper. Spoiler: more MDSAP content coming, like it or not..!
🇦🇪 UAE: Although it didn’t pass my filters, something in my gut tells me not to give up on it. I’d love to partner up with medtech experts and providers in that area.
This aligns well with the results from the poll: 52 votes, indicating predominant interest in "anglosphere" (32%) and "emerging" (30%), incl. LatAm.
Thanks to all who voted, commented and even called me to share their passionate insights!
Deep dive
The framework: three lenses, not one
We structured the analysis around three criteria, chosen because they reflect the real bottlenecks that founders face when entering new markets.
Regulatory alignment (MDSAP / MRAs). For a CE-marked or FDA-cleared device, the most efficient path into a new market is one that recognises or aligns with those approvals. The Medical Device Single Audit Program (MDSAP) is the primary international harmonisation vehicle, currently comprising the US (FDA), Canada (Health Canada), Brazil (ANVISA), Australia (TGA), and Japan (PMDA). Beyond MDSAP, a growing network of Mutual Recognition Agreements (MRAs) extends this regulatory compatibility further — most notably, the EU–Australia MRA, Israel's deep alignment with EU MDR, South Korea's MFDS harmonisation pathway, and GCC's increasing adoption of CE and FDA as reference standards.
Femtech readiness. This goes beyond market size. It asks whether the cultural, policy, and clinical infrastructure of a market is genuinely open to women's health innovation — or whether femtech companies will spend the first two years of market entry fighting the problem rather than solving it. We looked at startup density, clinical research activity, regulatory openness to digital health, and the status of women's health policy discourse in each candidate country.
ROI and medtech infrastructure. Commercial viability ultimately depends on whether a market can pay for solutions and scale them. This means evaluating healthcare financing structures (private payer vs. public reimbursement vs. out-of-pocket), digital health infrastructure, startup ecosystem depth, and the presence of credible distribution and partnership networks.
Only markets that scored meaningfully across all three lenses made the final shortlist. That intersection — regulatory alignment AND femtech readiness AND commercial infrastructure — is the filter that separated 8 markets from the 25+ we considered.
The 25-country universe and how it narrowed
We mapped approximately 25 countries across the three criteria, drawing on data from:
Institute for Economics & Peace Global Peace Index 2024 (163 countries) for geopolitical stability scoring
EF English Proficiency Index 2024 (116 countries) for accessibility
UN Population data 2024 for market size
World Bank ethnic fractionalization data for demographic heterogeneity (relevant to clinical research representativeness)
Vestbee, Dealroom, Speedinvest, Grand View Research, Startups Magazine for femtech market data by country
MDSAP member country documentation and publicly available MRA registers for regulatory alignment
Countries in only one or two circles of the Venn include genuinely interesting markets — Singapore (strong MDSAP alignment and infrastructure but too small for standalone commercial focus), India (exceptional femtech growth projected at 17.8% CAGR to 2030 per Grand View Research, but regulatory alignment is a work in progress), South Africa (emerging femtech ecosystem with strong policy momentum, but infrastructure gaps and geopolitical risk), China (significant commercial potential but regulatory and geopolitical friction makes it a poor fit for EU-anchored companies), and Kenya (one of Africa's most active femtech ecosystems, but not yet operationally viable for most European companies without a dedicated local strategy).
These are not dismissed markets — they are markets for a different risk appetite or a later stage.
The 8 that made the intersection
These are the markets that sat inside all three criteria simultaneously. For each, we break down the case across the three categories.
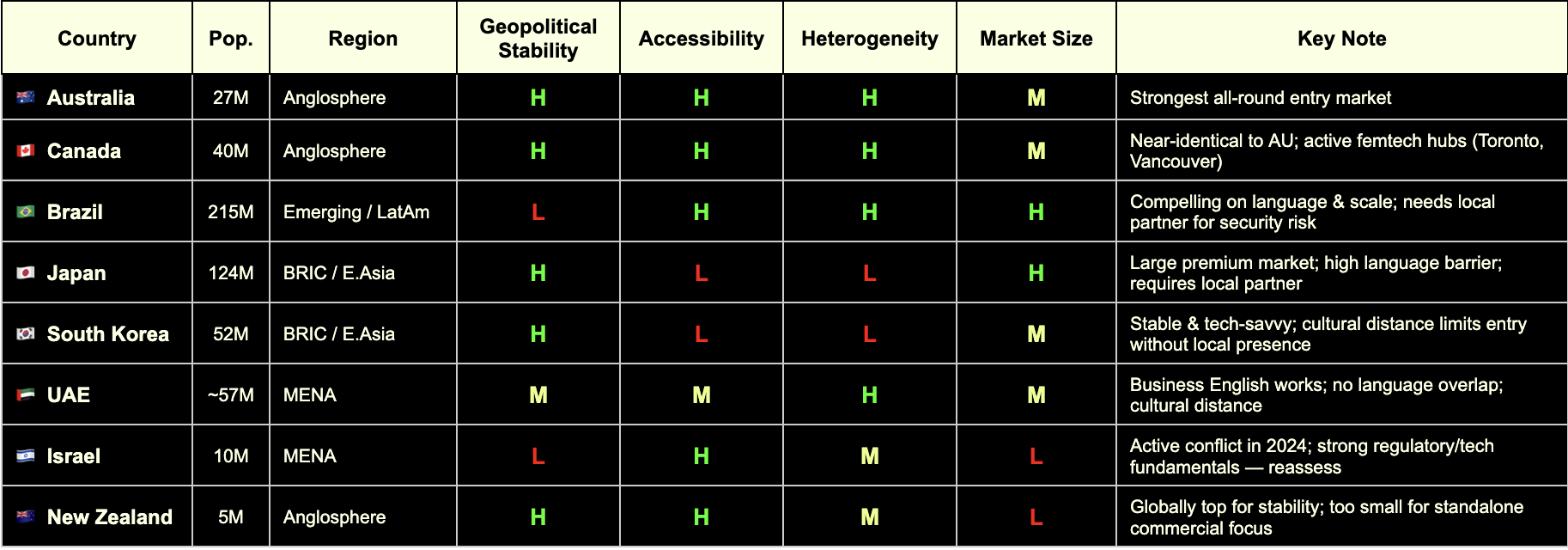
🇦🇺 Australia
Regulatory Alignment
MDSAP full member (TGA)
EU–Australia Mutual Recognition Agreement in place — CE mark directly supports TGA approval pathway
Strong alignment with ISO 13485 and IEC 62304 for software-based devices
Femtech Readiness
Mature digital health market with active government investment (Australian Digital Health Agency)
Women's health policy discourse is open and progressive; no significant cultural friction for femtech categories
Active medtech and healthtech startup ecosystem, particularly in Melbourne and Sydney
ROI & Infrastructure
Population ~27 million; 27% foreign-born (one of OECD's highest) — valuable for diverse clinical evidence
Mixed public/private payer system; growing private digital health reimbursement
English-speaking, Commonwealth-aligned, low operational complexity
GPI 2024: #17 — high geopolitical stability
🇨🇦 Canada
Regulatory Alignment
MDSAP founding member (Health Canada)
A single MDSAP audit covers Canada alongside the US, Brazil, Australia, and Japan — significant efficiency gain
Health Canada increasingly aligned with FDA on digital health device classification
Femtech Readiness
Toronto and Vancouver are active femtech hubs; Femtech Canada is an established national ecosystem
Strong clinical research infrastructure with diverse patient populations
Progressive women's health policy environment; high awareness and destigmatisation of femtech categories
ROI & Infrastructure
Population ~40 million; ~27% immigrant share — comparable diversity to Australia
Robust private and public payer landscape; provincial variation requires navigation
English and French markets; English operations are broadly sufficient for market entry
GPI 2024: #13 — highly stable
🇧🇷 Brazil
Regulatory Alignment
MDSAP full member via ANVISA — the most underutilised structural advantage for European companies entering LatAm
A single quality audit unlocks market access across all five MDSAP jurisdictions simultaneously
ANVISA has been modernising its device framework; registration timelines have improved significantly
Femtech Readiness
Growing digital health and femtech ecosystem, particularly in São Paulo and Rio de Janeiro
Large, young, highly digitally engaged female population
Significant unmet need in reproductive health, maternal health, and menstrual care — high receptivity to femtech solutions
ROI & Infrastructure
Population ~215 million — the largest in Latin America by a substantial margin
Highest ethnic fractionalization score globally (World Bank) — clinical data generated here is maximally representative
⚠️ GPI 2024: #131 — operational complexity is real; security considerations, contract risk, and local partner dependency must be factored in
Local partner infrastructure is a prerequisite, not an option
🇯🇵 Japan
Regulatory Alignment
MDSAP full member via PMDA (Pharmaceuticals and Medical Devices Agency)
PMDA is one of the most rigorous regulatory bodies globally; MDSAP audit significantly reduces duplicative burden
Regulatory pathway for digital health devices (SaMD) is well-defined
Femtech Readiness
One of the world's most advanced femtech markets by per-capita spend on menstrual health and reproductive technology
Strong clinical research infrastructure and data quality
⚠️ Cultural norms around women's health are evolving but remain conservative in some categories — market education investment required
ROI & Infrastructure
Population ~124 million; high healthcare spending and premium consumer health culture
High-margin market for validated, evidence-backed products
⚠️ EF English Proficiency Index: #92 out of 116 — significant language barrier; local partner or Japanese-speaking regulatory lead is non-negotiable
GPI 2024: #11 — very high stability
🇰🇷 South Korea
Regulatory Alignment
MFDS (Ministry of Food and Drug Safety) has progressively aligned with FDA and EU MDR frameworks
MDSAP compatibility pathway is expanding; not yet full membership but convergence is directional
Government actively promoting K-healthcare internationally — creating reciprocal openings for foreign market entry
Femtech Readiness
High digital health adoption and smartphone penetration; strong consumer appetite for health tracking and wearables
Growing awareness of women's health, particularly in fertility and menstrual health tech
Active startup ecosystem with increasing women's health focus
ROI & Infrastructure
Population ~52 million; advanced payer structures and tech scaling infrastructure
Government investment in digital health innovation is substantial
⚠️ EF EPI: #50 — business English limited outside Seoul; local presence required
GPI 2024: #46 — stable, with standard geopolitical considerations given peninsula context
🇮🇱 Israel
Regulatory Alignment
Israeli MOH operates with strong alignment to EU MDR and FDA standards — one of the most harmonised non-MDSAP markets globally
CE mark is a recognised reference standard; dual EU/FDA submission strategies are well-established
Deep bilateral ties with both EU and US regulatory bodies
Femtech Readiness
Exceptionally strong clinical research infrastructure; Israel produces a disproportionate number of medtech innovations relative to population
Highly developed VC and medtech ecosystem; femtech companies including HeraMed are globally recognised
High awareness and openness to women's health innovation; destigmatisation is advanced relative to regional peers
ROI & Infrastructure
Population ~10 million — small absolute market, but high income and high health spend per capita
English proficiency high (EF EPI: #51); business environment is highly accessible to European operators
Strong potential as a clinical trial site and R&D partnership hub, regardless of commercial scale
⚠️ GPI 2024: #155 — reflects active conflict situation as of the 2024 index; market entry decisions require live geopolitical risk assessment, not a static one. The underlying regulatory, clinical, and commercial infrastructure remains among the strongest in this cohort
🇦🇪 GCC (UAE & Saudi Arabia)
Regulatory Alignment
Neither is a formal MDSAP member, but UAE and KSA have adopted CE mark and FDA clearance as primary reference pathways for device registration
UAE MOHAP and Saudi SFDA increasingly streamlined; CE mark provides a strong starting point
GCC Standardization Organization (GSO) harmonisation creates some regional regulatory efficiency
Femtech Readiness
Saudi Arabia's Vision 2030 explicitly references women's empowerment and healthcare improvement — femtech is policy-aligned
UAE has the most open and internationally diverse women's health market in the region
Growing investor interest in femtech and digital health across the Gulf; GITEX Health and Arab Health are active deal-making forums
ROI & Infrastructure
UAE: ~89% of resident population is expatriate (UN data) — genuinely diverse patient population and cosmopolitan business environment
English is the functional operating language for business in both markets
Premium private-pay market; reimbursement structures favour high-value, evidence-backed products
⚠️ GPI 2024: UAE #53 (improved 31 places in 2024 — single largest improvement in the index); KSA requires ongoing monitoring
Cultural distance to women's health topics exists in some categories — local partnership and community navigation are important
🇳🇿 New Zealand
Regulatory Alignment
MDSAP full member (Medsafe)
Strong regulatory alignment with Australia (TGA); joint Trans-Tasman regulatory considerations apply
Simple, transparent regulatory environment — among the most accessible globally for device registration
Femtech Readiness
Progressive women's health policy environment; high health literacy and digital engagement
Maori and Pacific women's health is an active policy and research priority — relevant for inclusive femtech design and clinical evidence
English-speaking, low cultural friction across all standard femtech categories
ROI & Infrastructure
Population ~5 million — too small for standalone commercial focus
Best positioned as a clinical trial jurisdiction, regulatory proof-of-concept market, or Australasian stepping stone alongside Australia
GPI 2024: #4 — among the most geopolitically stable countries on earth
High income per capita; public system (Pharmac) for reimbursement is conservative but private market is accessible
What this means for women’s health startups
The eight markets are not interchangeable, and we are not suggesting a company should pursue all of them simultaneously. The practical read is this:
For a femtech company at the EU/US proof-of-concept stage, with limited expansion resources and no existing presence outside Europe, Australia and Canada are the lowest-friction first moves. They are MDSAP members, English-speaking, culturally accessible, and have enough market depth to justify the investment.
Brazil becomes compelling the moment you have a local partner or are prepared to build one. The MDSAP pathway is a structural advantage that is consistently underutilised by European companies, and a population of 215 million — with the highest demographic diversity of any market on this list — is a significant clinical research and commercial prize.
Japan and South Korea are high-reward markets that require deliberate localisation investment. The regulatory frameworks are accessible through MDSAP and MRA alignment; the human infrastructure needs to be built.
Israel and the GCC are specialist considerations — Israel for companies with strong clinical trial and research ambitions, the GCC for companies targeting the premium private-pay women's health segment and building into the Middle East and North Africa corridor.
New Zealand is genuinely valuable as a regulatory and clinical site — less so as a primary commercial target.
The 4th lens…
At Edge Compliance we want to keep serving US and EU market entries (EU as a continent, incl. UK and CH).
What this analysis taught us is where to put our next focus in terms of partnerships, network, learning, advocacy.
In order to avoid spreading ourselves too thinly, we applied some filters to narrow down the list of high potential regions to a smaller selection. The Edge Compliance lens considers:
Cultural accessibility
Market size
Demographic heterogeneity
Geopolitical stability
Criteria for market selection for Edge Compliance. Which are relevant to your business?
This analysis led us to choose the following areas of focus in addition to our existing focus and expertise:
🇧🇷 Brasil
🇦🇺 Australia
🇨🇦 Canada
🇦🇪 UAE
We trust this will help us help YOU with future-proof regulatory strategy and foresight that is ahead of trends.
References
Institute for Economics & Peace GPI 2024
EF English Proficiency Index 2024
UN World Population Prospects 2024
World Bank Ethnic Fractionalization Data
Vestbee Femtech Market Overview (2024)
Dealroom Femtech Report (2024)
Speedinvest Femtech Investment Analysis
Grand View Research India Femtech Market Outlook 2024–2030
MD+DI FemTech Analytics (2024)
Startups Magazine Southeast Asia Femtech Report
The Recursive CEE Femtech Analysis (2025)
Methodology note: This Deep Dive is based on my original LinkedIn post, reflecting my professional experiences and personal perspectives. Claude AI assisted in elaborating the topic into a broader article by integrating personal notes, literature research, fact-checking and deeper insights on the topic. All analysis and regulatory perspectives are my own, and all content has been reviewed by me for accuracy.
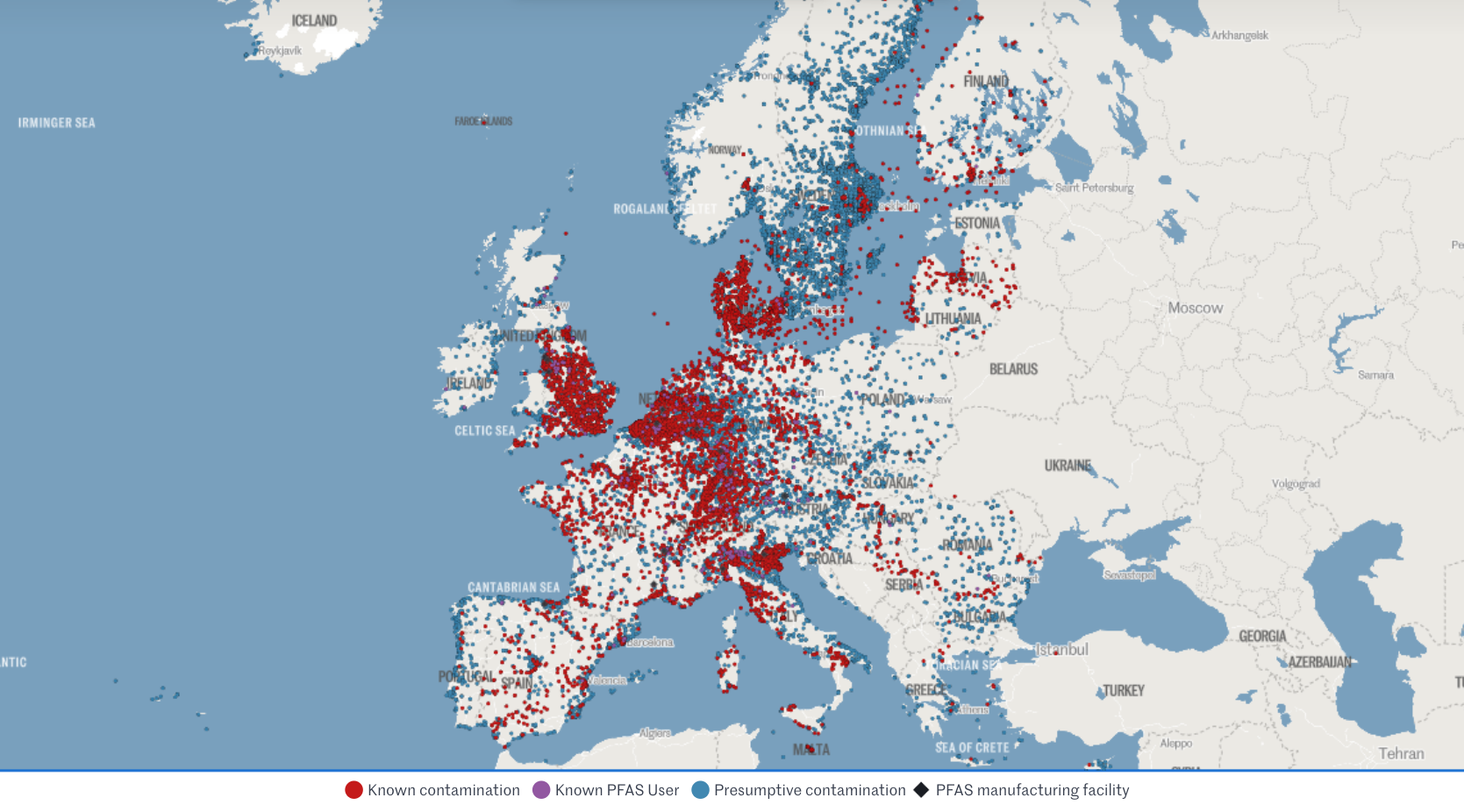
PFAS: wait-and-see Vs precautionary principle
When data is limited, do you default to safety or wait for proof of harm? This post explores the widening regulatory gap between the US and EU regarding PFAS - the "Forever Chemicals."
If you had to decide whether something is safe based on limited data, which way would you default?
Let's look at recent regulatory developments re "Teflon-like" chemicals (PFAS) in cosmetics and medical devices. Per- and polyfluoroalkyl substances (PFAS) are highly inert synthetic chemicals which makes them sought after for both everyday uses and specialist ones. However, they are so inert that biology cannot break them down. They persist in the environment and accumulate in creatures at the top of the food chain: us.
The regulatory approach to PFAS, also called Forever Chemicals, is another staggering example of the US vs. EU cultural divide.
U.S. wait-and-see approach
🇺🇸 Context: In 2024, FDA launched the Modernization of Cosmetics Regulation Act (MoCRA) which required registration of all cosmetics and listing of all their ingredients. This allowed FDA a fresh overview on PFAS' use in cosmetics, which inspired recent research.
🇺🇸 Research: A December 2025 report revealed that 51 types of PFAS are intentionally used in 1,744 cosmetic formulations in the US, commonly in makeup and even baby products.
🇺🇸 Conclusion: Due to a lack of critical toxicological data and acute toxicity, the safety of 76% of these compounds could not be definitively established. FDA deemed current evidence insufficient to justify a federal ban, opting instead for continued monitoring.
🇺🇸 Note: The FDA excluded environmental considerations and the assessment of unintentional degradation products, which are often the most harmful (e.g., PFOA and PFOS).
EU precautionary principle
🇪🇺 Context: The EU is already phasing out PFAS over concerns regarding long-term health effects and environmental contamination.
🇪🇺 Research: Rising concentrations in water streams and human blood (even in teenagers) are increasingly suspected to suppress the immune system and increase risks of cancer, infertility, thyroid dysfunction, and metabolic dysregulation.
🇪🇺 Conclusion: Action and monitoring stepped up at national and union level.
> This month, France has banned PFAS in all cosmetics (as well as clothing textiles and ski waxes).
> Yesterday, the European Environment Agency (EEA) kicked off a mandatory EU-wide program to systematically monitor PFAS in drinking water.
> Meanwhile, European Chemicals Agency (ECHA) is evaluating a proposal to ban 10,000 PFAS as a broad category, with stricter concentration limits (ppb levels) expected by October 2026.
🇪🇺 Note: The EU had already restricted all PFAS and even banned some under the REACh and the POPs regulations (which also impact allowed limits in medical devices under MDR).
Which side would you take? Personally, I’m leaning EU on this one.
Sources:
- FDA’s report
- EEA programme
- Forever pollution project (image credits)

FDA’s new guidance on general wellness
This post critiques the widening regulatory gap between the US and EU following the FDA's new wellness guidance, highlighting how lower barriers for bold health claims in the US may sacrifice essential quality drivers and complicate global strategies for startups.
Yesterday's release by FDA on wellness vs medical device leaves me with a bitter aftertaste. Why?
I'm usually enthusiastic about policies that lower the barrier to market entry for health products. I'm less enthusiastic about those that eliminate the quality drivers from it..
My main concerns under this guidance:
> General wellness products have no QMS requirement, especially digital ones. So when the guidance says you can now display biomarkers even with some disease reference as long as "the product has validated values" for those biomarkers, it doesn't really mean anything. How do they validate? According to what? Where? Claims get bolder and accountability weaker.
> We will see more products being Class IIa medical devices in EU (with QMS auditing and device file review) while facing zero expectations in the US as general wellness.
> The gap between EU and US regulatory approach gets wider. EU released a "similar" guidance in Sep 2025 emphasising the opposite, with increased focus on mechanism of action and technology rather than relying on claims only. US heads the other way, making it all the more complicated for us RA 🥴
> It will be harder for startups to design their product and strategies for the two main western markets simultaneously. They will be pushed even heavier towards wellness-first but in my experience they get easily stuck there.
> This bold approach may be (too) specific of this administration. Will it then outlive it? It is also clearly result from the WHOOP controversy, given the number of references to Blood Pressure measuring wrist-worn devices. Pretty solid legal and lobby teams there.
One example that puzzles me in particular is the one about glucose monitoring via "minimally invasive microneedle technology" for which FDA says they will apply enforcement discretion as a low risk device. Since I'm currently working on the biocompatibility testing requirements for a device that is hand held by doctors using gloves (👀), I cannot help but finding it unfair towards the rest of the sector.
So I hope you will excuse my slightly less upbeat post this time.
I'm generally excited about the expansion of the definition and agree with the rationale of most of the examples provided.
I'm curious to see what it will mean for international harmonisation and for the opportunities it will open for my clients at this interface!

MDR/IVDR proposal for simplication
This post highlights the European Commission's groundbreaking proposal to overhaul and simplify the MDR and IVDR frameworks, promising more proportionate rules for low-risk devices, reduced administrative burdens for SMEs, and a modern, digital-first approach to medtech regulation in the EU.
12 hours ago the European Commission published THE MOST AWAITED AND CRUCIAL DEVELOPMENT IN A DECADE: its proposal for simplification of the MDR and IVDR. 👏
Alert: it is still only a proposal, albeit official, which has been submitted to the European Parliament and the Council, but will need to go through the ordinary legislative procedure to become binding Union law.
From a first diagonal read, what struck my attention:
🎉 More room for Class I devices, incl software (THANK YOU!)
🎉 Simplified interaction with AI Act
🎉 Codified instruments for open dialogue on classification and access to expert panels
🎉 Easier "equivalence" concept including use of synthetic data,
🎉 Lower NB fee structure for SMEs
🎉 Extended reporting timelines and validity of certificates
🎉 Reduced scope of surveillance audits and conformity assessment
🎉 Built-in flexibility for public health emergencies, breakthrough/orphan devices (i.e. life-threatening, rare, untreated diseases), supply-chain disruptions
Interestingly, but unsurprisingly, it proposes additional requirements for cybersecurity conformity and reporting (beyond what qualifies as medically "serious").
I will share more details of how this would impact specifically medical device startups especially in digital health and femtech.
While it is still ONLY A PROPOSAL, it is sign that EU is listening and actively working to "make [the current rules] easier, faster and more effective and further promote competitiveness, innovation and a high-level of patient safety in this key sector"
We're excited to follow the development of the legislative decision-making process and wait eagerly for the change of an era this (or its variants that will result) will bring to the European medtech sector!

What can we learn from… Canada?
This post explores the "Canadian Technology Accelerator" model for international expansion, sharing insights from a bilingual FemTech panel in Paris on how high-potential startups - like PCOS-focused mentee Élan Healthcare - can leverage diplomatic networks and local mentorship to navigate global regulatory compliance.
What am I doing sitting in a bilingual English-French panel at the Embassy of Canada | Ambassade du Canada in Paris??
Talking femtech regulatory compliance trends (in English) while listening (mostly in live-translated French) to the perspectives of brilliant entrepreneurs, investors, researchers and diplomats!
This would be for my series of "What can we learn from... Canada?"
The Canadian Trade Commission runs the Canadian Technology Accelerators | Accélerateurs technologiques canadiens, a programme to support Canadian startups to expand to other markets. By collaborating with global Canadian embassies they provide eligible startups with local mentorship, contacts and partnerships to boost their growth.
As part of this, I had the privilege to mentor Élan Healthcare Inc. run by Pari (Parvaneh) Saharkhiz, MD, MBA, a doctor turned founder and manufacturer of supplements especially designed to tackle the nutritional imbalances that are often root to PCOS and infertility. Around 10% of women are affected by PCOS, 70% go undiagnosed, and even those who have it diagnosed struggle to find treatment. Check them out: https://elanhealthcare.ca/
Grateful for the invitation to Trade Commissioner Frederic Chieux and Fiona Thwaites. A pleasure to sit on the panel with collaborator, friend and amazing host Erica Perrier, PhD, MS, CSCS as well as great copanelists Régine Brielle Juliette Mauro Andrea Guest Andréa Saragoussi Keshiv Kaushal - thanks for sharing your knowledge.
Greatest success to the impressively advanced startups in the mentee cohort Cogni Cosm Medical Emovi Juno Technologies™ Mino Care My Normative LoOoP SYNG Pharmaceuticals Inc, I look forward to staying in touch!

At Swiss Medtech 2025
Swiss Medtech events never disappoint!
Key learnings from attending yesterdays session in sunny Bern (inside a stunning casino!):
1️⃣ US tariffs and lower FDA capacity are discouraging EU/CH startups from going US-first, but there are clever best-practices to work around them.
2️⃣ EU's gap between numbers in MDR applications and certifications is widening in unsustainable ways due to a poor EU-wide governance model for medtech, and how this needs fixing ASAP.
3️⃣ Switzerland is working out creative legal basis to be an attractive alternative (e.g. to fast-track FDA medical devices and to modernise its regulatory framework faster than the EU can)
4️⃣ Emerging markets (e.g. Saudi Arabia) get devices to market 6 months faster than traditional markets, meaning their patients get better outcomes, HCPs get better education, and the healthcare system innovates exponentially faster.
Grateful to Bernhard Bichsel and Sandra Item from ISS AG, Integrated Scientific Services, Daniel Delfosse, Eva von Mühlenen, LL.M., from Sidley Austin LLP, Glenda C. Marsh from Johnson & Johnson MedTech for putting together such an inspiring and informative afternoon!

The WHOOP saga
WHOOP ’s current FDA row is properly binge-worthy. Material for the next Lincoln Lawyer season on Netflix?
But until then, some personal reflections on why it matters for digital health and wearables.
This season’s hottest episodes:
🎞️ Ep. 1 : WHOOP launches Blood Pressure Insights (BPI) as a Wellness feature but claiming medical grade insights.
🎞️ Ep. 2 : FDA’s surveillance picks it up and issues a Warning Letter (made public with exceptional urgency) arguing against the medical disclaimers given the “inherent association” of BP with the diagnosis of hypo/hypertension,
🎞️ Ep. 3 : WHOOP refuses to pull the feature and takes it public/political, meeting with RFK Jr and attacking FDA’s integrity on social media.
I get it, it’s tough to live on the line. Enjoying the aura of “medical-grade” without the burden is the dream of many, but it's getting harder. I’ve been there with multiple startups, and deeply empathise with some of the operational and financial challenges they faced in getting that balance right - often in absence of clear guidelines.
But now: guidance is there, WHOOP already has an FDA-cleared ECG feature (i.e. a QMS) and likely the budget... then why not route the BPI feature under their existing regulated org? Whether from the start or in response to the warning. How is taking up this massive fight a better strategy?
In smaller cases, it would ring a quality culture and integration issue. But in this one, it’s seems a fight on principle - while enjoying the extra PR of being the torch bearer for the freedom of wearables worldwide.
Meanwhile, Hilo by Aktiia quietly secures BP clearance with medical indication for its bracelet without the fuss. 👀
If you’re in the borderline medical space, this is a defining moment.
➡️ Disclaimers may be shorter-lived than ever, careful if you’re relying on those.
➡️ Not all companies are the WHOOP or SPACEX of the ton. Don’t assume this aggressive strategy would work for you, play smart yes, but sustainable. PR and legal repercussions can be devastating for fundability.
➡️ Hire QARA professionals who know how to navigate the redlines vs the negotiables of borderline products.
As Blythe Karow put it in her BEAUTIFUL long read on this story:
“The art lies in reading between the lines and addressing the specific compliance issues rather than fighting fundamental regulatory doctrine.”
Meanwhile, in real life: A friend told me "my sleep/stress score from my watch is looking weird... am I sick??". Familiar? Apparently, WHOOP had an internal policy in place during COVID that employees should stay home if their score was lower than a certain threshold - they either had the virus or could easily get it. If this is how we use these tools, what's so bad in providing assurance of quality and accuracy in the first place?
Only time will tell.. For now, pass the popcorn 🍿